Variational Quantum Eigensolver (VQE)
למודול זה, הסטודנטים צריכים סביבת Python פעילה, ואת הגרסאות האחרונות של החבילות הבאות מותקנות:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
להגדרה והתקנה של חבילות אלה, ראו את המדריך התקנת Qiskit. להרצת משימות על מחשבי קוונטום אמיתיים, הסטודנטים יצטרכו להגדיר חשבון IBM Cloud, לפי השלבים במדריך הגדרת חשבון IBM Cloud שלך.
מודול זה נבדק ועשה שימוש של כ-8 דקות בזמן QPU. זה אומדן, והשימוש בפועל שלך עשוי להשתנות.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
מבוא
מאז פיתוח המודל המכני-קוונטי בתחילת המאה ה-20, מדענים הבינו שאלקטרונים אינם עוקבים אחרי מסלולים קבועים סביב גרעין האטום, אלא קיימים באזורי הסתברות הנקראים אורביטלים. אורביטלים אלה מתאימים לרמות אנרגיה ספציפיות ודיסקרטיות שאלקטרונים יכולים לתפוס. אלקטרונים שוהים באופן טבעי ברמות האנרגיה הנמוכות ביותר הזמינות, הידועות כמצב היסוד. אולם, אם אלקטרון קולט מספיק אנרגיה, הוא יכול לקפוץ לרמת אנרגיה גבוהה יותר ולהיכנס למצב מעורר. מצב מעורר זה הוא זמני, והאלקטרון ישוב בסופו של דבר לרמת אנרגיה נמוכה יותר, ויפלוט את האנרגיה שנקלטה, לרוב בצורת אור. תהליך בסיסי זה של קליטה ופליטת אנרגיה חשוב להבנת האינטראקציה בין אטומים ויצירת קשרים.
כאשר אטומים מתחברים ליצירת מולקולות, האורביטלים האטומיים שלהם משתלבים ליצירת אורביטלים מולקולריים. סידור רמות האנרגיה של האלקטרונים בתוך אורביטלים מולקולריים אלה קובע את תכונות המולקולה המתקבלת ואת חוזק הקשרים הכימיים. למשל, ביצירת מולקולת מימן () משני אטומי מימן נפרדים, האלקטרון מכל אטום תופס אורביטלים אטומיים. כשהאטומים מתקרבים זה לזה, אורביטלים אטומיים אלה חופפים ומשתלבים ליצירת אורביטלים מולקולריים חדשים — אחד עם אנרגיה נמוכה יותר (אורביטל קושר) ואחד עם אנרגיה גבוהה יותר (אורביטל אנטי-קושר). שני האלקטרונים, אחד מכל אטום מימן, יעדיפו לתפוס את אורביטל הקישור בעל האנרגיה הנמוכה, מה שיוביל ליצירת קשר קוולנטי יציב המחזיק את מולקולת ביחד. הפרש האנרגיה בין האטומים המופרדים לבין המולקולה שנוצרה, ובפרט אנרגיית האלקטרונים באורביטלים המולקולריים, קובע את יציבות ותכונות הקשר.
בחלקים הבאים נחקור תהליך יצירת המולקולה, תוך התמקדות במולקולת . נשתמש במחשב קוונטום אמיתי, בשילוב עם טכניקות אופטימיזציה קלאסיות, כדי למצוא את האנרגיה של תהליך פשוט אך בסיסי זה. ניסוי זה יספק הדגמה מעשית של כיצד ניתן ליישם חישוב קוונטי לפתרון בעיות בכימיה חישובית, ויספק תובנות לגבי תפקידה של אנרגיית האלקטרונים.
VQE - אלגוריתם קוונטי וריאציוני לבעיות ערכים עצמיים
טכניקות קירוב בכימיה - עקרון הווריאציה וסט הבסיס
תרומותיו של ארווין שרדינגר למכניקת הקוונטים אינן מוגבלות להצגת מודל אלקטרוני חדש; ביסודו, הוא ייסד את מכניקת הגלים על ידי פיתוח המשוואה המפורסמת של שרדינגר התלויה בזמן:
כאן, הוא אופרטור ההמילטוניאן, המייצג את האנרגיה הכוללת של המערכת, ו- היא פונקציית הגל המכילה את כל המידע על המצב הקוונטי של המערכת. (הערה: הוא הנגזרת הכוללת לפי זמן, ואנו לא כוללים כאן במפורש את ערך האנרגיה העצמית .)
אולם, ביישומים מעשיים רבים — כמו קביעת רמות האנרגיה המותרות של אטומים ומולקולות — אנחנו משתמשים במשוואת שרדינגר הבלתי-תלויה בזמן (משוואת ערך אנרגיה עצמי), הנגזרת מהצורה התלויה בזמן בהנחת מצב יציב. מצב יציב הוא מצב קוונטי שבו צפיפות ההסתברות למציאת חלקיק בנקודה נתונה במרחב אינה משתנה עם הזמן.
בצורה זו, מייצג את ערך האנרגיה העצמי המתאים למצב הקוונטי . ההמילטוניאן כולל תרומות אנרגיה שונות, כגון האנרגיה הקינטית של אלקטרונים וגרעינים, הכוחות המושכים בין אלקטרונים לגרעינים, והכוחות הדוחים בין אלקטרונים.
פתרון משוואת ערך האנרגיה העצמי מאפשר לנו לחשב את רמות האנרגיה הקוונטיות של מערכות אטומיות ומולקולריות. אולם, עבור מולקולות, פתרון מדויק הוא קשה מכיוון שפונקציית הגל , המתארת את הפיזור המרחבי של האלקטרונים, היא מורכבת ורב-ממדית.
כתוצאה מכך, מדענים משתמשים בטכניקות קירוב כדי לקבל פתרונות מעשיים ומדויקים. בעבודה זו, נתמקד בשתי שיטות מרכזיות:
-
עקרון הווריאציה
שיטה זו מקרבת את פונקציית הגל ומכוונת אותה כדי להתקרב ככל האפשר לאנרגיית המטרה, בדרך כלל אנרגיית מצב היסוד של המערכת. הרעיון המרכזי מאחורי עקרון הווריאציה הוא פשוט:
- אם אנחנו מנחשים פונקציית גל ("פונקציית ניסיון"), האנרגיה המחושבת ממנה תהיה תמיד שווה או גבוהה מאנרגיית מצב היסוד () של המערכת.
- על ידי כוונון פרמטרים בפונקציית הניסיון, , נוכל לקבל קירוב טוב יותר ויותר לאנרגיית מצב היסוד.
- הדיוק שלה תלוי מאוד בבחירת פונקציית הניסיון . פונקציית ניסיון שנבחרה רע עלולה להוביל לאומדן אנרגיה הרחוק מדיוק.
-
קירוב סט הבסיס
שיטת הקירוב השנייה מגיעה בשלב בניית פונקציית הגל — גישת סט הבסיס. בכימיה קוונטית, פתרון משוואת שרדינגר במדויק עבור מולקולות הוא כמעט בלתי אפשרי. במקום זאת, אנחנו מקרבים את פונקציית הגל המורכבת והרב-אלקטרונית על ידי בנייתה מפונקציות מתמטיות פשוטות ומוגדרות מראש. סט הבסיס הוא בעצם אוסף של פונקציות מתמטיות ידועות אלה, ממורכזות בדרך כלל על האטומים במולקולה, המשמשות כאבני בניין לייצוג צורתם והתנהגותם של האלקטרונים במערכת. חשבו על זה כמו ניסיון לשחזר פסל מפורט רק עם אוסף לבני LEGO סטנדרטיות — ככל שיש לנו יותר סוגים וגדלים של לבנים (ככל שסט הבסיס גדול יותר), כך נוכל לקרב את הצורה המקורית בדיוק רב יותר.
פונקציות הבסיס מונעות לעיתים קרובות מהפתרונות האנליטיים של מערכות פשוטות כמו אטום המימן, ולובשות צורות כמו פונקציות גאוסיאניות או מסוג סלייטר, אם כי הן עדיין קירובים. במקום לעבוד עם האורביטלים המולקולריים המלאים "המדויקים" תיאורטית אך בלתי ניתנים לטיפול, אנחנו מבטאים אותם כשילוב לינארי (סכום עם מקדמים) של פונקציות הבסיס הללו. שיטה זו ידועה כגישת קומבינציה לינארית של אורביטלים אטומיים (LCAO) כאשר פונקציות הבסיס דומות לאורביטלים אטומיים. על ידי אופטימיזציה של המקדמים בשילוב לינארי זה, נוכל למצוא את פונקציית הגל והאנרגיה המקורבות הטובות ביותר בתוך מגבלות סט הבסיס הנבחר.
- ככל שנכלול יותר פונקציות בסט הבסיס, כך הקירוב טוב יותר, אך זה בא במחיר של מאמץ חישובי גבוה יותר.
- סט בסיס קטן מספק אומדן גס, בעוד שסט בסיס גדול נותן תוצאות מדויקות יותר על חשבון דרישת משאבי חישוב רבים יותר.
לסיכום, כדי להפוך חישובים לאפשריים ולהפחית את העלות החישובית, אנחנו משתמשים בעקרון הווריאציה על ידי קירוב פונקציית הגל, מה שמפחית את המורכבות החישובית ומאפשר אופטימיזציה איטרטיבית למזעור אנרגיה. בינתיים, גישת סט הבסיס מפשטת חישובים על ידי ייצוג אורביטלים אטומיים כשילוב של פונקציות מוגדרות מראש, במקום לפתור פונקציית גל רציפה ישירות.
בדוק את ההבנה שלך
שקול את פונקציית הגל לניסיון כאשר הוא קבוע נרמול ו- הוא פרמטר ניתן לכוונון.
(a) נרמל את פונקציית הגל לניסיון על ידי קביעת כך ש- .
Answer
לנרמול פונקציית הגל הנתונה לניסיון:
השתמש באינטגרל הגאוסי:
הגדר ואז קבל:
(b) חשב את ערך הציפייה של ההמילטוניאן הנתון על ידי כאשר , המתאים לפוטנציאל אוסילטור הרמוני פשוט.
Answer
ההמילטוניאן עבור אוסילטור הרמוני הוא:
ערך ציפייה של האנרגיה הקינטית
לקיחת הנגזרת השנייה:
לפיכך:
שימוש בתוצאות אינטגרל גאוסי סטנדרטיות:
ערך ציפייה של האנרגיה הפוטנציאלית
שימוש ב:
נקבל:
ערך ציפייה של האנרגיה הכוללת
(c) השתמש בעקרון הווריאציה כדי למצוא את האופטימלי על ידי מזעור .
Answer
אופטימיזציה של לאנרגיה מינימלית
נגזור
פתרון:
הצבת ב-:
שמתאים לאנרגיית מצב היסוד המדויקת של האוסילטור הרמוני הקוונטי.
VQE (Variational Quantum Eigensolver)
ה-Variational Quantum Eigensolver (VQE) היא השיטה הראשית שבה נשתמש לחקר תהליך , וכאן נבחן מה זה VQE וכיצד הוא עובד. אבל קודם נעצור ונסתכל על דבר אחד חשוב מאוד דרך שאלת הבדיקה.
בדוק את ההבנה שלך
אם כבר יש לנו כל כך הרבה אסטרטגיות לבעיות כימיות, אז למה אנחנו צריכים מחשב קוונטום? ומה המטרה של שימוש בשני מחשבים קוונטיים וקלאסיים ביחד?
Answer
למחשוב קוונטי יש סיכוי לחולל מהפכה בכימיה על ידי התמודדות עם בעיות שמחשבים קלאסיים מתקשים בהן בשל הסקלאציה האקספוננציאלית של מצבים קוונטיים. ריצ'רד פיינמן ציין בפרסום את ההבחנה שכדי לדמות את הטבע, גם החישובים חייבים להיות קוונטיים [מקור 1].
למשל, דימוי קפאין עם סט הבסיס הפשוט ביותר (STO-3G) ידרוש ביטים, הרבה יותר מהמספר הכולל של כוכבים ביקום הנראה () [מקור 2]. מחשב קוונטי יכול לתאר את האורביטלים האלקטרוניים של קפאין עם 160 qubits.
מחשבים קוונטיים מעבדים באופן טבעי אינטראקציות קוונטיות באמצעות סופרפוזיציה ושזירה, המספקות דרך מבטיחה לאפשר סימולציות מולקולריות מדויקות. יתר על כן, אנחנו יכולים לשלב את היתרונות של מחשבים קוונטיים (סימולציית אלקטרונים) ומחשבים קלאסיים (עיבוד מקדים/לאחר של נתונים, ניהול תהליך האלגוריתם, אופטימיזציה, וכן הלאה). אלה צפויים לשפר את גילוי חומרים, עיצוב תרופות וחיזוי תגובות, ולהפחית ניסויי ניסוי-וטעייה יקרים. [מקור 3][מקור 4]
אם אתה רוצה לדעת למה צריך מחשבים קוונטיים לבעיות כימיות ולמה להשתמש גם במשאבי מחשוב קוונטיים וגם קלאסיים, בדוק את המאמרים הבאים:
עכשיו בוא נחזור ל-VQE.
VQE משלב את כוחם של מחשבים קוונטיים עם מחשבים קלאסיים, ובאופן בסיסי משתמש בעקרונות וריאציוניים כדי לקבל את אנרגיית מצב היסוד של המערכת. כדי להבין VQE, קודם פרקו אותו לשלושה חלקים:
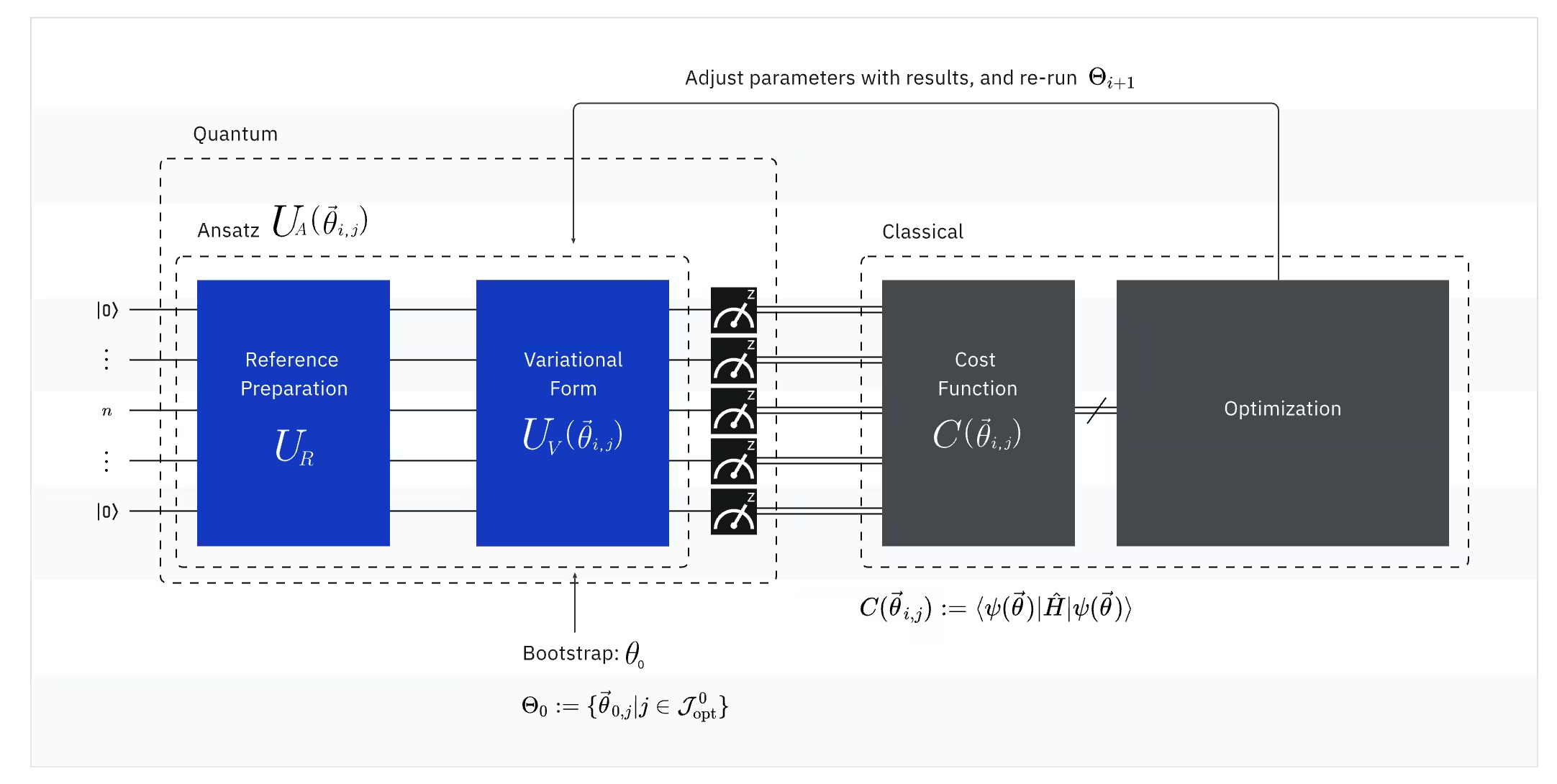
(קוונטי) גודל ניתן למדידה: ההמילטוניאן המולקולרי (אנרגיית מולקולה)
ב-VQE, ההמילטוניאן המולקולרי/אטומי הוא גודל ניתן למדידה, כלומר ניתן למדוד את ערכו דרך ניסוי. המטרה שלנו היא למצוא את האנרגיה הנמוכה ביותר האפשרית (אנרגיית מצב היסוד) של המולקולה. לשם כך, אנחנו משתמשים במצב קוונטי לניסיון, שנוצר על ידי Circuit קוונטי מפרמטרים (ansatz). אנחנו מודדים את הגודל הניתן למדידה ומבצעים אופטימיזציה על המצב הקוונטי עד שנגיע לאנרגיה הנמוכה ביותר האפשרית.
סט הבסיס המשמש עבור ההמילטוניאן המולקולרי קובע את מספר ה-Qubits הנדרשים ומשפיע ישירות על דיוק ה-VQE. בחירת סט הבסיס הנכון היא קריטית לאיזון בין יעילות ודיוק. כדי לפשט חישובים מבלי לשנות את סט הבסיס, ניתן להשתמש באסטרטגיות כמו הטלת סימטריה וצמצום מרחב פעיל. למולקולות רבות יש צורות סימטריות (כמו פרפר או פתית שלג), כלומר כמה חלקים מתנהגים באותו אופן. במקום לחשב הכול בנפרד, אנחנו יכולים להתמקד רק בחלקים ייחודיים, לחסוך משאבים קוונטיים, ובכך למנף סימטריה. בצמצום מרחב פעיל, אנחנו מתחשבים רק באורביטלים החשובים, שכן לא כל האלקטרונים משפיעים משמעותית על אנרגיית המולקולה. אלקטרונים קרובים לגרעין נשארים בעיקר ללא שינוי, בעוד שאחרים משפיעים על הקישור. על ידי יישום שיטות אלה, ניתן לייעל את ה-VQE תוך שמירת הדיוק.
לאחר שנקבל המילטוניאן מולקולרי באמצעות סט הבסיס והאסטרטגיות המתאימות לעיל, עלינו להפוך המילטוניאן זה לאחד המתאים למחשבים קוונטיים. מיפוי בעיות לאופרטורי פאולי יכול להיות מסובך למדי. זה נכון במיוחד בכימיה קוונטית, שעובדת עם חלקיקים בלתי ניתנים להבחנה (אלקטרונים), מכיוון ש-Qubits הם ניתנים להבחנה. לא נכנס לפרטי המיפויים כאן, אך אנחנו מפנים אותך למשאבים הבאים. דיון כללי על מיפוי בעיה לאופרטורים קוונטיים ניתן למצוא ב-Quantum computing in practice. דיון מפורט יותר על מיפוי בעיות כימיה לאופרטורים קוונטיים ניתן למצוא ב-Quantum chemistry with VQE.
עבור מודול זה, נספק לך את ההמילטוניאנים המתאימים (של qubit אחד) עבור ו- כדי שנוכל להתמקד בשימוש במחשב הקוונטי. המילטוניאנים אלה של qubit אחד מוכנים על ידי שימוש בסט הבסיס STO-6G ו-מיפוי ג'ורדן-ויגנר, שהוא המיפוי הישיר ביותר עם הפרשנות הפיזיקלית הפשוטה ביותר, מכיוון שהוא ממפה את התפוסה של אורביטל ספין אחד לתפוסה של qubit אחד. כמו כן, השתמשנו ב-טכניקת צמצום qubit באמצעות סימטריה של ההמילטוניאן, המשתמשת בדפוסים כיצד תפוסות ספין מתנהגות כדי לצמצם את מספר ה-Qubits. עבור מולקולת , אנחנו מניחים שהמרחק בין שני אטומי המימן הוא 0.735 .
(קוונטי) Ansatz: פונקציית הגל לניסיון (כיצד לבנות מצב קוונטי ראשוני עם Circuit קוונטי)
עבור VQE, ה-ansatz (רבים: ansätze) מורכב משני מרכיבים מרכזיים. הראשון הוא הכנת מצב ראשוני, שמגדירה את מצב ה-Qubit על ידי החלת Gates קוונטיים ללא פרמטר וריאציוני. המרכיב השני הוא ה-Circuit הקוונטי המפרמטרי, Circuit קוונטי מיוחד עם פרמטרים ניתנים לכוונון, בדומה לחוגות ברדיו. פרמטרים אלה ישמשו את האופטימייזר הקלאסי — החלק האחרון — כדי לעזור לנו להגיע למצב היסוד הטוב ביותר האפשרי.
בחלק עקרון הווריאציה, למדנו שאיכות המצב לניסיון משפיעה על איכות תוצאות האלגוריתם הווריאציוני. זה אומר שבחירת ansatz טוב חשובה ב-VQE. שוב, זהו נושא עשיר ומורכב. לא נכסה את סוגי ה-ansatz השונים או את מקורותיהם כאן. אם אתה מעוניין ללמוד עוד על Circuits קוונטיים מפרמטרים ו-ansatz, תוכל לחקור את השיעור Ansatz ו-variational form מקורס תכנון אלגוריתם וריאציוני, המספק הסברים ודוגמאות מפורטים של ansätze.
מכיוון שנשתמש בהמילטוניאן של qubit אחד במודול זה, אנחנו צריכים Circuit קוונטי מפרמטרי של qubit אחד כ-ansatz. נראה שלושה סוגים של ansätze של qubit אחד בחלק הבא. נשווה אותם ונדון בשיקולים מרכזיים בבחירת ansatz.
(קלאסי) אופטימייזר: כוונון עדין של ה-Circuit הקוונטי
ברגע שהמחשב הקוונטי מודד את האנרגיה של הגודל הניתן למדידה מה-ansatz, הפרמטרים של ה-ansatz וערך האנרגיה נשלחים לאופטימייזר הקלאסי לכוונון. תהליך אופטימיזציה זה מבוצע על מחשב קלאסי, בדרך כלל באמצעות חבילות מדע כלליות כמו SciPy.
האופטימייזר הקלאסי מתייחס לאנרגיה הנמדדת כפונקציית עלות. בבעיות אופטימיזציה, פונקציית עלות (הנקראת לעיתים גם פונקציית מטרה) היא פונקציה מתמטית המודדת כמה "טוב" פתרון מסוים. המטרה של האופטימייזר היא למצוא את קבוצת הפרמטרים שממזערת פונקציית עלות זו. בהקשר של מציאת אנרגיית מצב היסוד של מולקולה, האנרגיה עצמה משמשת כפונקציית העלות — אנחנו רוצים למצוא את הפרמטרים עבור ה-Circuit הקוונטי שלנו ("הפתרון" שלנו) המניבים את האנרגיה הנמוכה ביותר האפשרית. האופטימייזר הקלאסי משתמש בערך האנרגיה הנמדדת זה (העלות) וקובע את קבוצת הפרמטרים הממוטבים הבאה עבור ה-ansatz הקוונטי. פרמטרים מעודכנים אלה נשלחים לאחר מכן בחזרה ל-Circuit הקוונטי, והתהליך חוזר על עצמו. עם כל איטרציה, האופטימייזר הקלאסי מכוונן את הפרמטרים כדי לנסות ולהפחית את האנרגיה (למזער את פונקציית העלות) עד שיתמלא קריטריון התכנסות מוגדר מראש, ובאופן אידיאלי מבטיח שתימצא האנרגיה הנמוכה ביותר האפשרית (המתאימה למצב היסוד של המולקולה לאותו מרחק קשר וסט בסיס).
ישנן אסטרטגיות אופטימיזציה רבות הניתנות על ידי חבילות מדע כמו SciPy. תוכל למצוא עוד בשיעור לולאות אופטימיזציה של קורס תכנון אלגוריתם וריאציוני. כאן נשתמש ב-COBYLA (Constrained Optimization BY Linear Approximations), אלגוריתם אופטימיזציה מתאים לנופי אנרגיה מורכבים. בפרט, COBYLA לא מנסה לחשב גרדיאנט של הפונקציה הנחקרת; זה נקרא אופטימייזר ללא גרדיאנט. דמיין שאתה מנסה למצוא את הפסגה הגבוהה ביותר ברכס הרים בעיניים עצומות. מכיוון שאינך יכול לראות את כל הנוף, אתה נוקט בצעדים קטנים לכיוונים שונים, תוך בדיקה אם אתה עולה או יורד. COBYLA עובד בצורה דומה — הוא נע דרך מרחב הפרמטרים, בוחן ערכים שונים, ומשפר בהדרגה את התוצאה עד שהוא מוצא את הטוב ביותר.
עכשיו אתה מוכן לבצע חישוב VQE. לשם כך, נסה את שאלת הבדיקה למטה, המסכמת את התהליך הכולל.
בדוק את ההבנה שלך
מלא את החסרים במונחים הנכונים כדי להשלים את סיכום תהליך ה-VQE, ולאחר מכן לחץ לבדוק את תשובותיך.
VQE הוא אלגוריתם קוונטי וריאציוני, המשלב את כוחם של (1) ________ ומחשוב קלאסי, המשמש למציאת (2) __________ של מולקולה. התהליך מתחיל בהגדרת (3) __________, המייצג את האנרגיה הכוללת של המערכת ומשמש כגודל ניתן למדידה במדידות קוונטיות. לאחר מכן, אנחנו מכינים (4) __________, Circuit קוונטי עם פרמטרים ניתנים לכוונון המייצג את פונקציית הגל לניסיון של המולקולה. פרמטרים אלה מותאמים אופטימלית באמצעות (5) __________, אלגוריתם קלאסי המכוונן פרמטרים באיטרטיביות כדי למזער את האנרגיה הנמדדת. בדיון לעיל השתמשנו ב-(6) __________, אשר מחדד את פרמטרי ה-ansatz מבלי שהוא זקוק לחישובי נגזרות. התהליך נמשך עד שאנו מגיעים ל-(7) __________, כלומר מצאנו את האנרגיה הנמוכה ביותר האפשרית של המולקולה.
בנק מילים:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Answer
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
חישוב אנרגיית מצב היסוד של אטום מימן עם VQE
עכשיו, בוא נשתמש במה שלמדנו כדי לחשב את אנרגיית מצב היסוד של אטום מימן. לאורך המודול, נשתמש במסגרת עבודה לחישוב קוונטי הנקראת "Qiskit patterns", שמחלקת תהליכי עבודה לשלבים הבאים:
- שלב 1: מיפוי קלטים קלאסיים לבעיה קוונטית
- שלב 2: אופטימיזציה של הבעיה להרצה קוונטית
- שלב 3: הרצה באמצעות פרימיטיבים של Qiskit Runtime
- שלב 4: עיבוד לאחר מכן וניתוח קלאסי

בדרך כלל נלך בצעדים האלה.
בוא נתחיל בטעינת כמה חבילות נחוצות, כולל פרימיטיבים של Qiskit Runtime. כמו כן נבחר את המחשב הקוונטי הפחות עמוס שזמין לנו.
יש קוד למטה לשמירת האישורים שלך בשימוש הראשון. הקפד למחוק את המידע הזה מהמחברת לאחר שמירתו לסביבתך, כדי שהאישורים שלך לא ישותפו בטעות כשתשתף את המחברת. ראה הגדר את חשבון IBM Cloud שלך ו-אתחל את השירות בסביבה לא מהימנה לקבלת הנחיות נוספות.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
התא שלמטה יאפשר לך לעבור בין שימוש בסימולטור או בחומרה אמיתית לאורך המחברת. אנחנו ממליצים להריץ אותו עכשיו:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
שלב 1: מיפוי הבעיה ל-Circuit קוונטיים ואופרטורים
אנחנו מתחילים את חישוב ה-VQE שלנו בהגדרת ההמילטוניאן עבור מולקולת המימן () במרחק קשר ספציפי. ההמילטוניאן הזה מייצג את האנרגיה הכוללת של המערכת במונחים של אופרטורי qubit, לאחר שהופק ומופה ממערכת המולקולה באמצעות נוהל סטנדרטי: 1) שימוש בבסיס STO-6G (אוסף ספציפי של פונקציות מתמטיות המשמשות לקירוב אורביטלים אלקטרוניים), 2) יישום מיפוי ג'ורדן-ויגנר (טכניקה לתרגום אופרטורים פרמיוניים המתארים אלקטרונים לאופרטורי qubit), ו-3) ביצוע הפחתת qubit באמצעות פריטט של ההמילטוניאן לפישוט הבעיה.
כפי שהסברנו קודם, אנרגיות מצב היסוד המחושבות תלויות מאוד בבחירת הבסיס ובגיאומטריה המולקולרית (כמו מרחק הקשר). עבור תצורה ספציפית זו ולאחר ביצוע הטרנספורמציות הללו, ההמילטוניאן של qubit המתקבל הוא פשוט:
כאן, מייצג את אופרטור הזהות ו- מייצג את אופרטור פאולי-Z, הפועל על qubit יחיד. המקדמים נגזרים מהאינטגרלים המחושבים באמצעות בסיס STO-6G במרחק הקשר המסוים הזה עם טרנספורמציה נכונה.
עם ההמילטוניאן הזה מוגדר, אנחנו יכולים להשתמש ב-VQE כדי לחשב את אנרגיית מצב היסוד שלו. כדאי להשוות את אנרגיית מצב היסוד המחושבת שלנו לערכים צפויים. עבור אטום מימן (H) בודד ומבודד, אנרגיית מצב היסוד היא בדיוק -0.5 Hartree (בהיעדר אפקטים רלטיביסטיים). בוא נחשב את אנרגיית מצב היסוד המדויקת של ההמילטוניאן הספציפי של ה-Qubit שלנו כפי שהוגדר לעיל ונשווה אותה לערכים ידועים רלוונטיים.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
לאחר מכן, אנחנו צריכים Circuit קוונטי עם פרמטרים, ansatz, להכנת פונקציית גל ניסיונית עבור מצב היסוד. המטרה היא למצוא את הפרמטרים שממזערים את ערך הציפיה של האנרגיה . בחירת ה-ansatz היא קריטית מכיוון שהיא קובעת את קבוצת המצבים הקוונטיים האפשריים ש-Circuit שלנו יכול להכין. "ansatz טוב" הוא כזה שגמיש מספיק לייצוג מצב קרוב מאוד למצב היסוד האמיתי של ההמילטוניאן שאנחנו חוקרים, אך לא כל כך מורכב שהוא דורש יותר מדי פרמטרים או Circuit עמוק מדי עבור מחשבים קוונטיים נוכחיים.
כאן, ננסה שלושה ansätze חד-Qubit שונים כדי לראות איזה מהם מספק "כיסוי" טוב יותר של המצבים הקוונטיים האפשריים ש-Qubit יחיד יכול להיות בהם. ה"כיסוי" מתייחס לטווח המצבים הקוונטיים ש-Circuit ה-ansatz יכול לייצר על ידי שינוי הפרמטרים שלו.
נשתמש בשלושה ansätze המבוססים על שילובים שונים של Gate סיבוב חד-Qubit:
- ansatz עם Gate סיבוב חד-ציר אחד: ansatz זה משתמש בסיבובים סביב ציר יחיד בלבד (). על כדור בלוך, זה מתאים לתנועה לאורך מעגל ספציפי בלבד. זהו הגמיש פחות ומכסה קבוצה מוגבלת של מצבים.
- שני ansätze עם Gate סיבוב דו-ציר: ansätze אלה משלבים סיבובים סביב שני צירים שונים ( ו-). זה מאפשר לנו להגיע לחלק גדול יותר של כדור בלוך, בהשוואה לסיבוב חד-ציר.
על ידי השוואת תוצאות VQE שהתקבלו עם שלושת ה-ansätze הללו, נוכל לראות כיצד הגמישות וכיסוי מרחב המצבים של ה-ansatz משפיעים על יכולתנו למצוא את אנרגיית מצב היסוד האמיתית של ההמילטוניאן הפשוט שלנו. ansatz גמיש יותר בעל פוטנציאל למצוא קירוב טוב יותר, אך עשוי להיות קשה יותר לאופטימייזר הקלאסי.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
עכשיו, בוא נייצר 5000 מספרים אקראיים לכל פרמטר ונשרטט את ההתפלגות של מצבים קוונטיים אקראיים, שנוצרו על ידי שלושת ה-ansätze עם הפרמטרים האקראיים הללו. אפשר לחשוב על הפרמטרים האלה כמו סיבובים סביב צירים שונים על פני כדור. כדי לראות את התפלגות המצב הקוונטי, נשתמש בכדור בלוך, כדור תלת-ממדי המציג את מצב qubit יחיד. כל נקודה על הכדור מייצגת מצב אפשרי של ה-Qubit, כאשר הקטבים הצפוני והדרומי הם כמו ה"0" וה"1" הקלאסיים, אך ה-Qubit יכול גם להיות בכל מקום באמצע, ולהציג תכונות קוונטיות מיוחדות כמו סופרפוזיציה. ראשית, הכן את הפונקציות הנחוצות לשרטוט כדור בלוך התלת-ממדי והכן 5000 פרמטרים אקראיים.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
בוא נראה איך ה-ansatz הראשון שלנו עובד.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
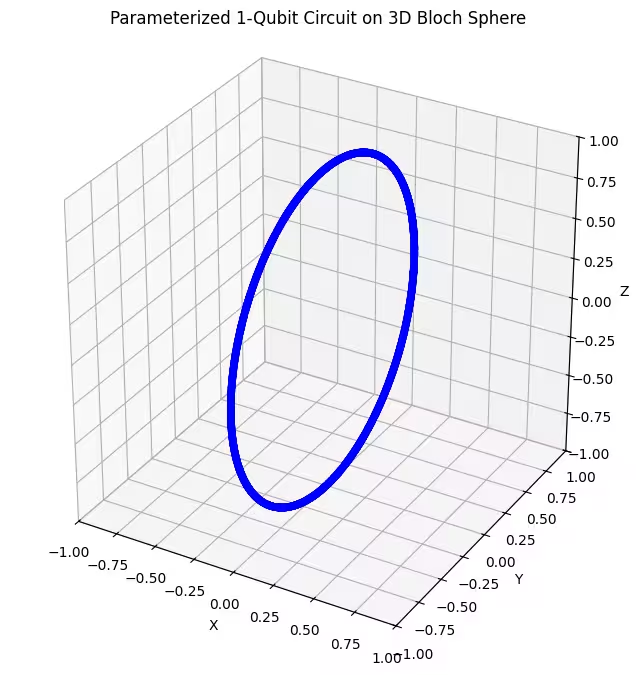
אנחנו יכולים לראות שה-ansatz הראשון שלנו מחזיר מצבים קוונטיים המפוזרים בצורת טבעת על כדור בלוך. זה הגיוני, כי נתנו ל-ansatz פרמטר סיבוב יחיד בלבד. הוא לכן יכול לייצר רק מצבים המסובבים סביב ציר אחד. התחלה מהנקודה וסיבוב סביב ציר אחד תניב תמיד טבעת. אחר כך בוא נבדוק את ה-ansatz השני שלנו, שיש לו שני Gate סיבוב ניצבים - Rx ו-Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
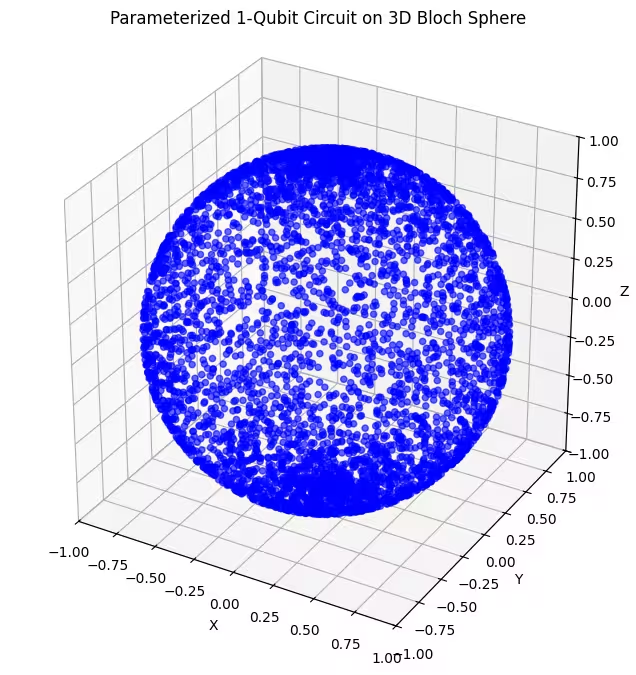
כאן, אנחנו יכולים לראות שה-ansatz השני שלנו מכסה חלק גדול יותר של כדור בלוך - אך שים לב שהנקודות מרוכזות יותר סביב הקטבים ומפוזרות יותר סביב קו המשווה. עכשיו הגיע הזמן לבדוק את ה-ansatz האחרון שלנו.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
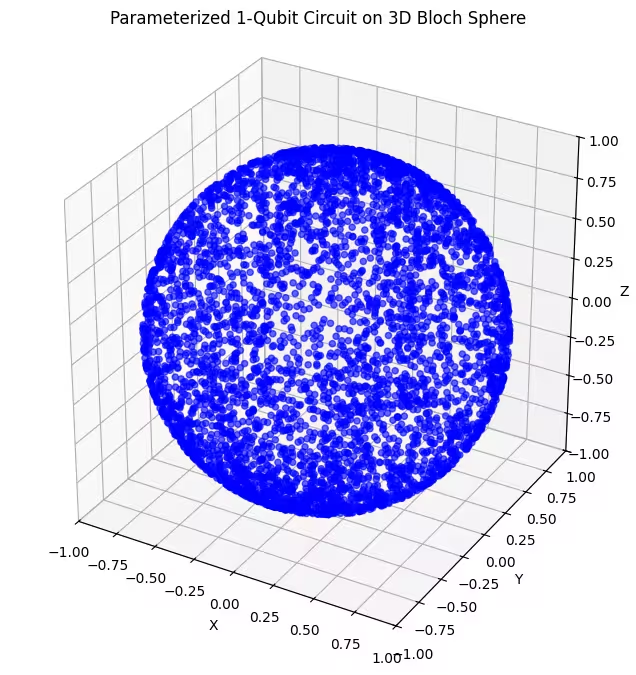
כאן אפשר לראות מצבים קוונטיים מפוזרים באופן אחיד יותר שנוצרו על ידי ה-ansatz האחרון שלנו.
כפי שצוין, הדבר הכי טוב לעשות הוא לרכוש ידע על מצב היסוד שאתה מחפש ולהשתמש ב-ansatz שמתאים היטב לבדיקת מצבים קרובים למצב היסוד הזה. לדוגמה, אם ידענו שמצב היסוד שלנו נמצא ליד קוטב, אולי היינו בוחרים ב-ansatz 2. לפשטות, נישאר עם ansatz 3, שבודק באופן אחיד את כדור בלוך כולו.
עכשיו שבחרנו את ה-ansatz שלנו, בוא נשרטט את ה-Circuit.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

שלב 2: אופטימיזציה עבור חומרת היעד
כשמריצים חישוב על מחשב קוונטי אמיתי, לא אכפת לנו רק מהלוגיקה של ה-Circuit הקוונטי. אכפת לנו גם מדברים כמו אילו פעולות יכולות להתבצע על ידי אותו מחשב קוונטי ספציפי, ואיפה על המחשב הקוונטי נמצאים ה-Qubitים שאנחנו משתמשים בהם. האם הם ממש ליד זה? האם הם רחוקים זה מזה? לכן, הצעד הבא הוא לשכתב את ה-Circuit שלנו באמצעות Gate שטבעיים למחשב הקוונטי שנשתמש בו, תוך התחשבות בפריסת ה-Qubitים. ניתן לעשות זאת על ידי transpilation - לאחר תהליך זה, אפשר לראות את ה-ansatz הפשוט שלנו הומר לקבוצה שונה של Gate, וה-Qubitים המופשטים שלנו ימופו ל-Qubitים פיזיים על מחשב קוונטי אמיתי.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

אפשר לראות שה-Gate rx, rz של ה-ansatz שלנו הומרו לסדרה של Gate rz, sx, שהם ה-Gate הטבעיים של ה-Backend שלנו. כמו כן, אפשר לראות ש-q0 שלנו ממופה עכשיו ל-Qubit הפיזי החמישי. אנחנו גם צריכים למפות את ההמילטוניאן שלנו בהתאם לשינויים הללו, כפי שמוצג בקוד הבא:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
שלב 3: הרצה על חומרה אמיתית
עכשיו הגיע הזמן להריץ את ה-VQE שלנו על QPU אמיתי. לשם כך, קודם כל אנחנו צריכים פונקציית עלות לתהליך האופטימיזציה, שמחשבת את ערך הציפייה של ההמילטוניאן עם מצב קוונטי שנוצר על ידי ה-ansatz. אל תדאגו! אתם לא צריכים לקודד הכל בעצמכם. הכנו פונקציה לכך, וכל מה שאתם צריכים לעשות הוא להריץ את התא למטה.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
לבסוף, אנחנו מכינים פרמטרים ראשוניים עבור ה-ansatz ותהליך האופטימיזציה שלו. אפשר פשוט להשתמש באפסים או בערכים אקראיים. בחרנו פרמטרים ראשוניים למטה, אבל אתם מוזמנים להוסיף או להסיר הערות בתא כדי לדגום פרמטרים באופן אקראי ואחיד מ-0 עד .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
כל הכבוד! סיימתם בהצלחה את הניסוי הראשון שלכם בכימיה קוונטית. אנחנו רואים הפרש בין אנרגיית מצב היסוד המדויקת של ההמילטוניאן לבין שלנו, אבל מאחר שהשתמשנו בטכניקת מיתון שגיאות ברירת מחדל (שמתקנת שגיאות קריאה), ההפרש קטן. זהו התחלה מצוינת!
הערה: אפשר לקבל תוצאה טובה יותר על ידי הגדרת רמת מיתון שגיאות באמצעות resilience_level. ערך ברירת המחדל הוא 1, ואם מגדירים ערך גבוה יותר, ייעשה שימוש בזמן QPU רב יותר אך ייתכן שיוחזרו תוצאות טובות יותר.
שלב 4: עיבוד לאחר מעשה
הגיע הזמן להסתכל על איך עבד האופטימייזר הקלאסי שלנו. הרץ את התא למטה וראה את דפוס ההתכנסות.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

התחלנו עם ערך ראשוני טוב למדי, כך שקיבלנו ערך סופי טוב בסה"כ ב-10 צעדים. אפשר לראות פסגות גדולות וקטנות, וזו התכונה האופיינית של אופטימייזר COBYLA — הוא חוקר את המרחב כאילו אינו יכול לראות את הנוף ומתאים את גדלי הצעד עם כל מדידה.
בדוק את ההבנה שלך
מה התצפית שלך? איזה חלק מהתהליך לעיל פתוח לשיפור כדי לקבל תוצאות קרובות יותר לערכים התיאורטיים, או קרובות יותר לאנרגיית מצב היסוד המדויקת של ההמילטוניאן? מה כדאי לשקול לשם כך?
Answer
הדבר הראשון לשקול הוא שינוי בסט הבסיסים המשמש לחישוב ההמילטוניאן של המולקולות. כפי שהוזכר קודם, אנרגיית מצב היסוד של אטום H היא -0.5 Hartree, כידוע, ובסיס STO-6G שבחרנו אינו מספיק כדי לגזור ערך זה בדייקנות.
בחירת סוג בסיס מורכב יותר מגדילה את מספר ה-Qubits שבהם משתמש ההמילטוניאן; לכן, אנחנו צריכים לבחור ansatz מורכב ומתאים יותר לבעיות כימיה.
הבא לאופטימיזציה הוא ניהול הרעש ב-QPU. טכניקות מיתון שגיאות מתקדמות יותר מניבות תוצאות טובות יותר אך עשויות לקחת יותר זמן לשימוש. כמו כן, כדאי לשקול כיצד shot_number משפיע על התוצאות.
לבסוף, ביצועי התכנסות טובים יותר ניתן להשיג גם על ידי ניסוי אופטימייזרים שונים.
חישוב אנרגיית מצב היסוד של מולקולת המימן עם VQE
עכשיו שהסתכלנו על התהליך הכולל של VQE באמצעות אטומי , נחשב את אנרגיית מצב היסוד של מולקולת בצורה מהירה יותר.
שלב 1: מיפוי הבעיה ל-Circuits ואופרטורים
כאן אנחנו גם מספקים לכם המילטוניאן חד-Qubit שמשתמש בבסיס STO-6G ובהמרת Jordan-Wigner, עם הפחתת qubit באמצעות סימטריה של ההמילטוניאן. שימו לב שהשתמשנו במרחק אטומי בין שני אטומי מימן של 0.735 .
שלא כמו בחישוב של אטום מימן יחיד (), כדי לחשב את מצב היסוד של מולקולת מימן (), אנחנו חייבים לשקול גם את הכוח הדוחה הפועל בין הגרעינים של שני אטומי המימן, בנוסף לאנרגיה הקשורה לאורביטלים האלקטרוניים. בשלב זה, נתן ערך זה כקבוע, ובפועל נחשב ערך זה בשאלת הביניים.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
שלב 2: אופטימיזציה עבור חומרת היעד
מאחר שמספר ה-Qubits שבהם משתמשים ה-VQE וההמילטוניאן הקודמים זהה ל-Backend שישמש להרצה, נשתמש ב-ansatz הקיים ובצורתו האופטימית.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
שלב 3: הרצה על חומרה אמיתית
עכשיו הגיע הזמן לבצע את החישובים על QPU אמיתי. כמעט הכל זהה, אך נשתמש בנקודת התחלה מתאימה להתאמה להמילטוניאן. כמו כן, בחלק האיטרטיבי, חלק מהגדרות ה-Estimator, המשמש לחישוב ציפיות ההמילטוניאן עבור ה-ansatz ב-QPU, יוגדרו מעט שונה מהחישובים הקודמים. נדון בשינוי זה עוד בשאלת הביניים.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
למרות ש-VQE מספק תיאורטית גבול עליון לאנרגיית מצב היסוד האמיתית, מימושים מעשיים על חומרה קוונטית אמיתית או על סימולציה רועשת, כמו גם הפשטות שנעשות בהכנת ההמילטוניאן (כמו סטי בסיסים או הפחתת qubit), עלולות להכניס שגיאות שלפעמים מביאות לאנרגיה מדודה הנמוכה מעט מהערך התיאורטי המדויק או ממסמך ייחוס מספרי ספציפי. למרות שיש כמה שגיאות, התוצאות נראות מספקות, במיוחד לאור מספר הצעדים הקטן. עכשיו, בואו נסיים את חישוב ה-VQE הזה על ידי הסתכלות על איך עבד האופטימייזר.
שלב 4: עיבוד לאחר מעשה
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

בדוק את ההבנה שלך
בואו נחשב את אנרגיית הדחייה הגרעינית של מולקולת , שכללנו כערך קבוע (0.71997 Hartree).

השתמש בחוק קולון ובמערכת יחידות אטומיות כדי לוודא שאתה מקבל את הערך ב-Hartree.
Answer
מאחר ששני גרעיני המימן טעונים חיובית, הם דוחים זה את זה בגלל הכוח האלקטרוסטטי. דחייה זו מתוארת על ידי חוק קולון:
כאשר הוא מטען הפרוטון, הוא מאפשרות הוואקום, ו- הוא המרחק בין שני הגרעינים, נמדד במטרים או ברדיוסי Bohr ביחידות ג'ול (J).
כדי לחשב אנרגיה זו ב-Hartrees, אנחנו צריכים להמיר את המשוואה לעיל למערכת יחידות אטומיות (AU). ב-AU, , ורדיוס Bohr () הוא 1 והופך לסקלת האורך הבסיסית ב-AU. עם פישוטים אלו, חוק קולון מצטמצם ל:
כאשר חייב להיות נמדד ברדיוסי Bohr ().
כדי להמיר את הפרדת הגרעינים הנתונה ב- ל-, אנחנו צריכים יחס המרה זה:
לכן הופך ל-.
לפיכך, אנרגיית הדחייה הגרעינית של הנתון היא
חישוב אנרגיית התגובה של
עכשיו בואו נשתמש במה שהשגנו! השתמשת ב-VQE, פותר ערכים עצמיים קוונטי וריאציוני, כדי לחשב את אנרגיית מצב היסוד של אטום ושל מולקולת . מה שנותר הוא להשתמש בערכים שחישבנו כדי לקבל את אנרגיית התגובה של התהליך .
אנרגיית תגובה היא שינוי האנרגיה שקורה כאשר חומרים מגיבים ויוצרים חומרים חדשים. דמיין שאתה בונה משהו: לפעמים צריך להשקיע אנרגיה (כמו לערום קוביות), ולפעמים אנרגיה משתחררת (כמו כדור שמתגלגל במורד הגבעה). בכימיה, תגובות הן או בולעות אנרגיה (אנדותרמיות) או משחררות אנרגיה (אקסותרמיות).
ניתן לחשב את אנרגיית התגובה של התהליך לפי הנוסחה הבאה:
על ידי הרצת התא למטה, בואו נראה את זה בצורה ויזואלית. כאן נשתמש בערך מצב היסוד המדויק של כל המילטוניאן, ונשווה את אנרגיית התגובה של הפתרון המדויק ושל תוצאות VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

כפי שמוצג באיור, למרות שיש כמה שגיאות, אנרגיית מצב היסוד המדויקת של ההמילטוניאנים ואנרגיית התגובה שחושבה בעזרת תוצאות VQE דומות, קרובות ל-0.2- Hartree.
ראוי לציין כאן שאנרגיית התגובה של תהליך זה היא שלילית, מה שאומר שאנרגיה משתחררת במהלך התהליך, והמולקולה שנוצרת בעלת אנרגיה נמוכה יותר משני אטומים בודדים. 6. סיכום
בואו נסכם את מה שלמדנו עד כה.
קודם כל הסתכלנו על שתי טכניקות קירוב חשובות הנחוצות לפתרון בעיות כימיה קוונטית: עקרון הווריאציה ובחירת ערכות הבסיס, שהן שתיהן בסיסיות ל-VQE. חקרנו את עקרון הווריאציה ביד, וחישבנו את אנרגיית מצב היסוד של האוסצילטור ההרמוני הפשוט.
לאחר מכן חקרנו את VQE, אלגוריתם נפוץ לחישוב אנרגיית מצב היסוד של מערכת קוונטית. הרצנו קוד לחישוב אנרגיות מצב היסוד של מימן אטומי () ושל מולקולת המימן (). בפרט, למדנו שיש צורך לקבל את ההמילטוניאן המולקולרי המתאים למערכת ולהמיר אותו לצורה שניתן להריץ על מחשב קוונטי. ראינו גם שה-ansatz, Circuit קוונטי עם פרמטרים, נדרש להכנת מצבי גלים קוונטיים ניסיוניים בתוך VQE, ודנו בחשיבות בחירת מבנה Circuit ה-ansatz המתאים. למדנו גם שVQE מסתמך על תהליך אופטימיזציה איטרטיבי תוך שימוש במחשב קלאסי, המנחה את ה-Circuit הקוונטי למצוא את מצב האנרגיה הנמוכה ביותר, וראינו כיצד התהליך מתכנס.
לבסוף, השתמשנו באנרגיות מצב היסוד המחושבות של ו- שהתקבלו דרך VQE כדי לחשב את אנרגיית התגובה עבור התהליך .
VQE הוא אלגוריתם קוונטי עוצמתי לטווח הקרוב, אך חשוב להיות מודעים למגבלותיו. הביצועים של VQE תלויים מאוד בבחירת ה-ansatz — מציאת ansatz שניתן להכין ביעילות ויכול לייצג במדויק את מצב היסוד האמיתי הופכת מאתגרת יותר עבור מולקולות גדולות ומורכבות יותר. יתר על כן, חומרה קוונטית נוכחית רגישה לרעש, מה שיכול להשפיע על דיוק תוצאות VQE, במיוחד עבור Circuits עמוקים יותר או מספרים גדולים יותר של qubits. למרות האתגרים האלה, VQE משמש כאלגוריתם בסיסי, ומחקר מתמשך חוקר שיטות וריאציוניות מתוחכמות יותר וטכניקות הפחתת שגיאות כדי לדחוף את גבולות האפשרי בכימיה קוונטית על מחשבים קוונטיים לטווח הקרוב. למשל, אלגוריתמים כמו Sample-based Quantum Diagonalization (SQD) מפותחים כיום, שמנצלים דגימות שהתקבלו מ-Circuits קוונטיים בשילוב עם אלכסוניזציה קלאסית בתת-מרחב כדי לשפר את הערכת האנרגיה ולטפל בחלק מהמגבלות שVQE מתמודד איתן, במיוחד בנוגע ליעילות המדידה ועמידות ברעש.
סקירה ושאלות
מושגים מרכזיים:
- אלגוריתם קוונטי וריאציוני הוא פרדיגמת חישוב שבה מחשב קלאסי ומחשב קוונטי עובדים יחד לפתרון בעיה.
- ב-VQE, אנחנו מתחילים עם ההמילטוניאן של המערכת שלנו וממפים אותו ל-Qubits להרצה על המחשב הקוונטי. אנחנו בוחרים Circuit קוונטי עם פרמטרים, ansatz, ועושים מדידות חוזרות, משנים את הפרמטרים של ה-ansatz, עד שמגיעים לערך האנרגיה הנמוך ביותר. החיפוש במרחב הפרמטרים נעשה בעזרת אופטימייזר קלאסי. כדי להשיג תוצאות טובות, יש לבחור ansatz טוב ואופטימייזר מתאים.
- אנרגיית תגובה היא שינוי האנרגיה הכולל בתגובה כימית, הנקבע על ידי ההפרש בין אנרגיית המגיבים לאנרגיית התוצרים.
נכון/לא נכון
- עקרון הווריאציה קובע שערך הציפייה של האנרגיה עבור כל פונקציית גל ניסיונית הוא תמיד גדול מאנרגיית מצב היסוד האמיתי או שווה לה.
- ערכת בסיס היא אוסף של פונקציות המשמשות לקירוב פונקציות גל קוונטיות.
- VQE הוא אלגוריתם קוונטי המשמש לפתרון מדויק של משוואת שרדינגר עבור המילטוניאן נתון.
- ב-VQE, Circuit קוונטי עם פרמטרים (ansatz) משמש להכנת פונקציות גל ניסיוניות.
- בחירת האופטימייזר ב-VQE (למשל, COBYLA, SPSA, או ADAM) אינה משפיעה על איכות התוצאה.
- ה-
Estimatorשל Qiskit משמש לחישוב ישיר של ערכי ציפייה של המילטוניאנים ב-VQE.
שאלות רב-ברירה:
- מה המטרה של המילטוניאן ב-VQE?
- א) ליצור מצבים קוונטיים אקראיים
- ב) לקבוע את האנרגיה של מצבים קוונטיים
- ג) לאופטמז Circuit קוונטי
- ד) ליצור שזירה
- מה המטרה העיקרית של אלגוריתם VQE?
- א) למצוא את אנרגיית מצב היסוד של ההמילטוניאן
- ב) ליצור שזירה בין qubits
- ג) לבצע את חיפוש גרובר
- ד) לשבור את הצפנת RSA
- כמה מצבים קוונטיים נוצרים ב-notebook זה להשוואת ה-ansatz?
- א) 100
- ב) 1000
- ג) 5000
- ד) 10,000
- מדוע נדרש אופטימייזר קלאסי ב-VQE?
- א) לביצוע מדידות קוונטיות
- ב) לעדכן פרמטרי ansatz כדי למזער אנרגיה
- ג) לשזור qubits
- ד) ליצור אקראיות קוונטית
- מדוע ה-ansatz מתוכנן להיות עם פרמטרים?
- א) כדי לאפשר הכנת מצב קוונטי
- ב) כדי לאפשר חיפוש במרחב רחב של מצבים קוונטיים
- ג) כדי לצמצם את מורכבות ה-Circuit
- ד) כדי למדוד ערכים עצמיים ישירות
- מה ההצהרה הנכונה ביותר לגבי בחירת ansatz טוב?
- א) ansatz חייב לייצר מצבים מפוזרים באופן שווה על כדור בלוך, אחרת הוא ייכשל.
- ב) ansatz צריך להיות מותאם למערכת שלך כדי לוודא שהוא יכול לייצר מצבים קרובים למצב היסוד.
- ג) ansatz צריך לייצר מצבים אקראיים באמצעות הפרמטרים הווריאציוניים שלו.
- ד) ansatz טוב יותר תמיד כולל יותר פרמטרים וריאציוניים.
(אופציונלי) נספח: עומס האופטימייזר לפי מורכבות ה-ansatz
VQE מתמודד עם כמה אתגרים ידועים[ref 6], והבאים קשורים למה שלמדנו למעלה.
- אתגרי בחירת ansatz
קיים אתגר מובנה בבחירת ה-ansatz הווריאציוני הנכון. ansätze בהשראת כימיה (כמו UCCSD) מספקים דיוק פיזי אך דורשים Circuits עמוקים, בעוד ansätze יעילים לחומרה הם Circuits רדודים יותר אך עשויים לחסור פרשנות פיזית. כמו כן, ansätze רבים מציגים פרמטרים וריאציוניים מיותרים שתורמים מעט לשיפור הדיוק אך מגדילים משמעותית את קושי האופטימיזציה.
- קשיי אופטימיזציה
נוף האופטימיזציה של VQE יכול לכלול אזורים שבהם גרדיאנטים פוחתים באופן אקספוננציאלי (רמות שטוחות — barren plateaus), מה שמקשה על אופטימייזרים קלאסיים לעדכן את הפרמטרים הווריאציוניים ביעילות. לשם כך, חוקרים ניסו להשתמש בסוגים שונים של אופטימייזרים — מבוססי גרדיאנט וחופשיים מגרדיאנט — אך שניהם מתמודדים עם אתגרים. אופטימייזרים מבוססי גרדיאנט סובלים מרמות שטוחות, בעוד שיטות חופשיות מגרדיאנט דורשות מספר רב של הערכות פונקציה.
- עומס האופטימייזר
אתגר ידוע נוסף הוא עומס האופטימייזר, שקשור לגודל הבעיה. ה-Circuits הקוונטיים הנדרשים ל-VQE גדלים בעומק ובמורכבות ככל שגודל הבעיה גדל; זה בדרך כלל גם מגדיל את מספר הפרמטרים לאופטימיזציה. תהליך האופטימיזציה הופך בלתי-ניתן לביצוע ככל שמספר הפרמטרים גדל, מה שמוביל להתכנסות איטית ולקשיים במציאת הפתרון האופטימלי.
כאן נבחן את האתגרים האלה על ידי שימוש ב-VQE עבור מולקולת , עם שני סוגים שונים של ansätze.
(הערה: זה עשוי לקחת יותר זמן QPU, אז אל תהסס להשתמש בסימולטור אם אין לך מספיק זמן.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
עכשיו בואו נריץ VQE עם נקודת התחלה של כולם אחד, עם מקסימום 20 צעדים, ונשווה את ההתכנסות של שני הריצות.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

הגרף למעלה ממחיש בבירור שתהליך האופטימיזציה של ה-ansatz עם יותר משתנים לוקח יותר זמן להגיע להתכנסות יציבה.
במקום להסתמך על Circuits פשוטים של qubit בודד ו-ansatz ישיר, מורכבות האופטימיזציה גדלה כאשר נדרשים Circuits קוונטיים גדולים יותר ו-ansätze בנויים במבנה מורכב יותר. זה מדגיש אתגר ידוע ב-VQEs: עומס האופטימייזר.
חוקרים ממשיכים לפתח מתודולוגיות מתקדמות שונות שיכולות להשתמש במחשבים קוונטיים לבעיות כימיה. ניתן לגשת למגוון חומרי לימוד ב-IBM Quantum Learning.
מקורות
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)