שיפור אמידת האנרגיה של המילטוניאן כימי עם SQD
במדריך זה נממש תבנית Qiskit שמראה כיצד לעבד בדיעבד דגימות קוונטיות רועשות כדי למצוא קירוב למצב היסוד של המילטוניאן כימי: מולקולת בשיווי משקל בבסיס 6-31G. נעקוב אחר גישת אלכסון קוונטי מבוסס דגימות לעיבוד דגימות שנלקחו ממעגל אנזץ קוונטי בן 36 qubit (במקרה זה, מעגל LUCJ). כדי להתחשב בהשפעת הרעש הקוונטי, נשתמש בטכניקת שחזור הקונפיגורציה.
ניתן לתאר את התבנית בארבעה שלבים:
- שלב 1: מיפוי לבעיה קוונטית
- יצירת אנזץ לאמידת מצב היסוד
- שלב 2: אופטימיזציה של הבעיה
- Transpile של האנזץ עבור ה-Backend
- שלב 3: ביצוע ניסויים
- שליפת דגימות מהאנזץ באמצעות ה-Primitive
Sampler
- שליפת דגימות מהאנזץ באמצעות ה-Primitive
- שלב 4: עיבוד תוצאות בדיעבד
- לולאת שחזור קונפיגורציה עצמית-עקבית
- עיבוד בדיעבד של כל הדגימות של מחרוזות הסיביות, תוך שימוש בידע מוקדם על מספר החלקיקים ותפוסת האורביטל הממוצעת שחושבה באיטרציה האחרונה.
- יצירה הסתברותית של אצוות משניות מהמחרוזות המשוחזרות.
- הטלה ואלכסון של ההמילטוניאן המולקולרי על כל תת-מרחב מדוגם.
- שמירת אנרגיית מצב היסוד המינימלית שנמצאה בכל האצוות ועדכון תפוסת האורביטל הממוצעת.
- לולאת שחזור קונפיגורציה עצמית-עקבית
לדוגמה זו, המילטוניאן של אלקטרונים מקיימי אינטראקציה לובש את הצורה הכללית:
/ הם אופרטורי היצירה/ההשמדה הפרמיוניים המשויכים לאיבר ה- של קבוצת הבסיס ולספין . ו- הם האינטגרלים האלקטרוניים חד-גופניים ודו-גופניים.
תהליך העבודה של SQD עם שחזור קונפיגורציה עצמית-עקבית מוצג בתרשים הבא.
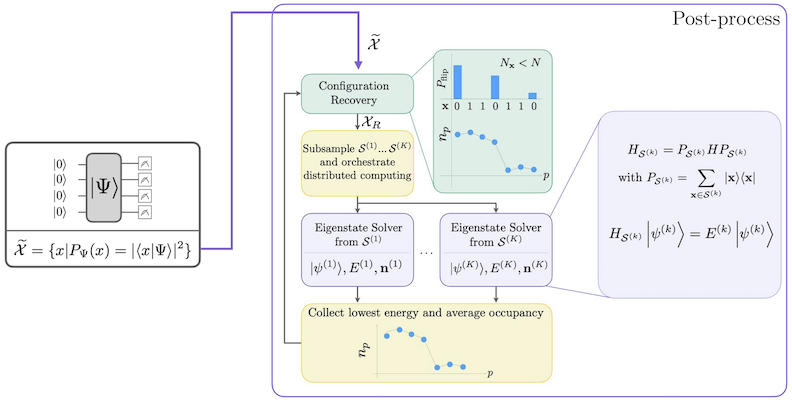
ידוע ש-SQD עובד היטב כאשר מצב הייגן המטרה הוא דליל: פונקציית הגל נתמכת על קבוצת מצבי בסיס שגודלה אינו גדל באופן מעריכי עם גודל הבעיה. בתרחיש זה, אלכסון ההמילטוניאן המוטל על תת-המרחב המוגדר על-ידי :
מניב קירוב טוב למצב הייגן המטרה. תפקיד המכשיר הקוונטי הוא לייצר דגימות של איברי בלבד. ראשית, מעגל קוונטי מכין את המצב במכשיר הקוונטי. נעשה שימוש בקידוד Jordan-Wigner. כתוצאה מכך, איברי בסיס החישוב מייצגים מצבי Fock (קונפיגורציות/דטרמיננטים אלקטרוניים). המעגל נדגם בבסיס החישוב, ומניב את קבוצת הקונפיגורציות הרועשות . הקונפיגורציות מיוצגות על-ידי מחרוזות סיביות. הקבוצה מועברת לאחר מכן לבלוק העיבוד הקלאסי בדיעבד, שבו נעשה שימוש בטכניקת שחזור הקונפיגורציה העצמית-עקבית. במסגרת SQD, תפקיד המכשיר הקוונטי הוא לייצר התפלגות הסתברות.
שלב 1: מיפוי הבעיה למעגל קוונטי
במדריך זה נקרב את אנרגיית מצב היסוד של מולקולת . ראשית, נגדיר את המולקולה ואת מאפייניה. לאחר מכן, ניצור אנזץ local unitary cluster Jastrow (LUCJ) (מעגל קוונטי) לייצור דגימות ממחשב קוונטי לצורך אמידת אנרגיית מצב היסוד.
ראשית, נגדיר את המולקולה ואת מאפייניה.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
לאחר מכן, ניצור את האנזץ. האנזץ LUCJ הוא מעגל קוונטי פרמטרי, ונאתחל אותו עם אמפליטודות t2 ו-t1 שהתקבלו מחישוב CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
נשתמש בחבילה ffsim ליצירת האנזץ ולאתחולו עם אמפליטודות t2 ו-t1 שחושבו לעיל. מכיוון שמולקולתנו בעלת מצב Hartree-Fock עם קליפה סגורה, נשתמש בגרסה המאוזנת-ספין של אנזץ UCJ, UCJOpSpinBalanced.
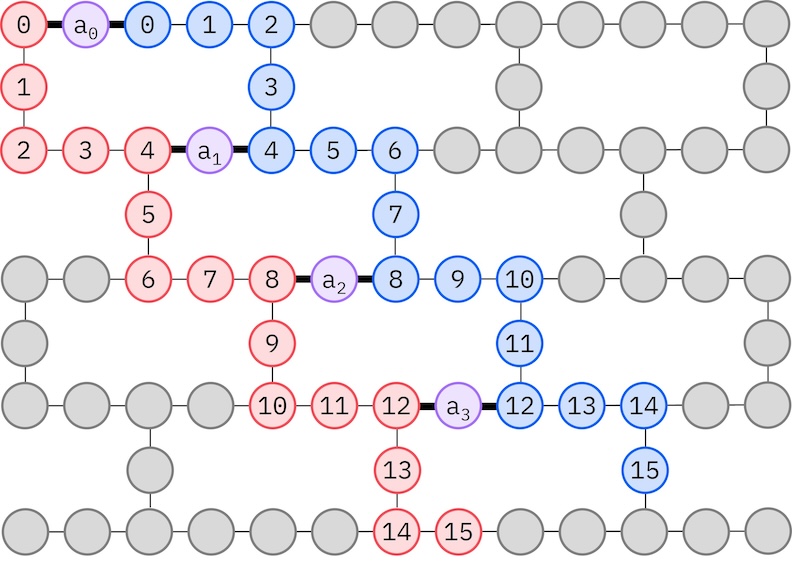
מכיוון שלחומרת IBM המטרה שלנו יש טופולוגיית hex-כבד, נאמץ את תבנית הזיג-זג לאינטראקציות qubit. בתבנית זו, אורביטלים (המיוצגים על-ידי qubit) עם אותו ספין מחוברים בטופולוגיית קו (עיגולים אדומים וכחולים) שבה כל קו לובש צורת זיג-זג בשל הקישוריות של hex-כבד של החומרה המטרה. שוב, בשל טופולוגיית hex-כבד, לאורביטלים עם ספינים שונים יש חיבורים בין כל אורביטל 4 (0, 4, 8, וכו') (עיגולים סגולים).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
שלב 2: אופטימיזציה של הבעיה
לאחר מכן, נבצע אופטימיזציה של המעגל שלנו עבור חומרה מטרה. עלינו לבחור את מכשיר החומרה שבו נשתמש לפני שנבצע אופטימיזציה של המעגל. נשתמש ב-Backend מדומה בת 127 qubit מ-qiskit_ibm_runtime כדי לחקות מכשיר אמיתי. כדי להריץ על QPU אמיתי, פשוט החלף את ה-Backend המדומה ב-Backend אמיתי. ראה במסמכי Qiskit IBM Runtime למידע נוסף.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
לאחר מכן, אנחנו ממליצים על השלבים הבאים לאופטימיזציה של האנזץ והפיכתו לתואם-חומרה.
- בחר qubit פיזיים (
initial_layout) מהחומרה המטרה בהתאם לתבנית הזיג-זג שתוארה לעיל. פריסת qubit בתבנית זו מובילה למעגל יעיל תואם-חומרה עם פחות Gate. - צור מנהל מעבר בשלבים באמצעות הפונקציה generate_preset_pass_manager מ-Qiskit עם הבחירה שלך של
backendו-initial_layout. - הגדר את שלב
pre_initשל מנהל המעבר בשלבים שלך ל-ffsim.qiskit.PRE_INIT. ffsim.qiskit.PRE_INITכולל מעברי Transpiler של Qiskit שמפרקים Gate לסיבובי אורביטל ולאחר מכן ממזגים את הסיבובים, מה שמביא לפחות Gate במעגל הסופי. - הרץ את מנהל המעבר על המעגל שלך.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
שלב 3: ביצוע ניסויים
לאחר אופטימיזציה של המעגל להרצה על חומרה, אנחנו מוכנים להריץ אותו על החומרה המטרה ולאסוף דגימות לאמידת אנרגיית מצב היסוד. מכיוון שיש לנו רק מעגל אחד, נשתמש במצב הרצת Job של Qiskit Runtime ונבצע את המעגל שלנו.
הערה: הגבנו בהערה את הקוד להרצת המעגל על QPU והשארנו אותו כעזר למשתמש. במקום הרצה על חומרה אמיתית במדריך זה, נייצר פשוט דגימות אקראיות הנשלפות מהתפלגות אחידה.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
שלב 4: עיבוד תוצאות בדיעבד
כעת, נריץ את אלגוריתם SQD באמצעות הפונקציה diagonalize_fermionic_hamiltonian. ראה את תיעוד ה-API להסברים על הארגומנטים לפונקציה זו.
הפותר הכלול ב-addon של SQD משתמש במימוש של PySCF לבחירת CI, ספציפית pyscf.fci.selected_ci.kernel_fixed_space. הדוגמה למטה מראה גם כיצד להעביר ארגומנטים מילוליים לפונקציה זו דרך הפותר הכלול. כאן אנחנו מעבירים את הארגומנט max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
כעת, נשרטט את התוצאות.
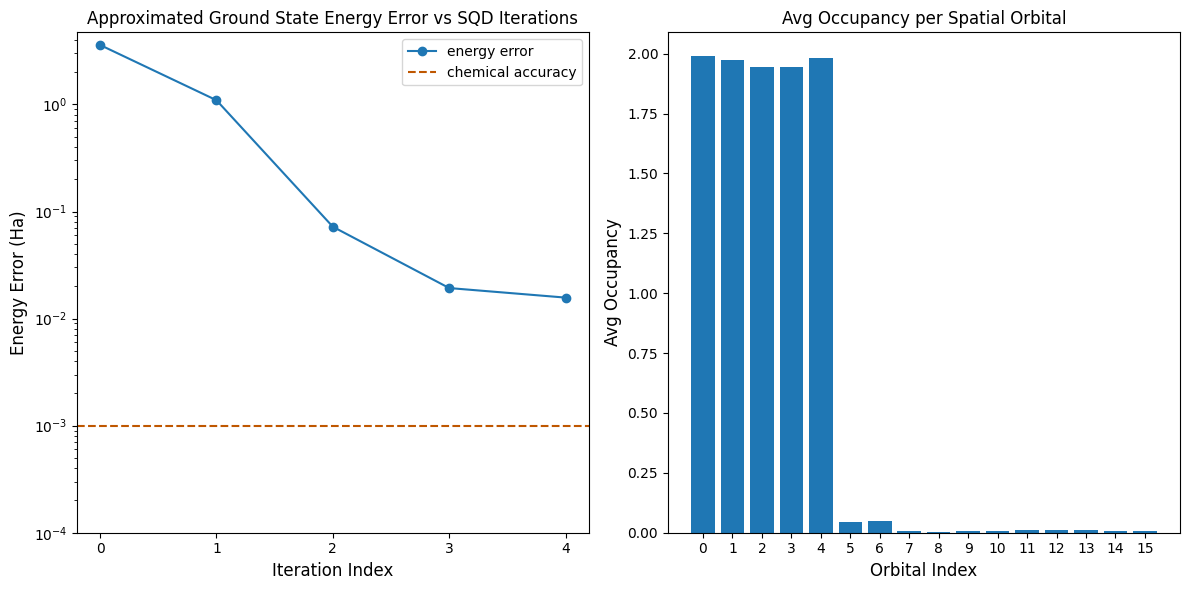
הגרף הראשון מראה שלאחר מספר איטרציות אנחנו מעריכים את אנרגיית מצב היסוד בטווח של ~16 mH (הדיוק הכימי מקובל בדרך כלל כ-1 kcal/mol 1.6 mH). זכור, הדגימות הקוונטיות בהדגמה זו היו רעש טהור. האות כאן מגיע מידע מוקדם על המבנה האלקטרוני וההמילטוניאן המולקולרי.
הגרף השני מראה את תפוסת האורביטל המרחבי הממוצעת לאחר האיטרציה האחרונה. אנחנו יכולים לראות שגם האלקטרונים עם ספין-מעלה וגם אלה עם ספין-מטה תופסים את חמשת האורביטלים הראשונים בהסתברות גבוהה בפתרונות שלנו.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha