HI-VQE Chemistry - פונקציית Qiskit מאת Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
ראה את ה-API reference
פונקציות Qiskit הן תכונה ניסיונית הזמינה רק למשתמשי IBM Quantum® Premium Plan, Flex Plan, ו-On-Prem (דרך IBM Quantum Platform API) Plan. הן נמצאות בסטטוס מהדורה מקדימה ועשויות להשתנות.
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
סקירה כללית
בכימיה קוונטית, בעיית המבנה האלקטרוני מתמקדת במציאת פתרונות למשוואת שרדינגר האלקטרונית — פונקציות גל קוונטיות המתארות את ההתנהגות של אלקטרוני המערכת. פונקציות גל אלו הן וקטורים של אמפליטודות מרוכבות, כאשר כל אמפליטודה מתאימה לתרומתה של תצורת אלקטרונים אפשרית.
מצב היסוד הוא פונקציית הגל של אנרגיה מינימלית של המערכת ויש לו חשיבות מיוחדת בחקר מערכות מולקולריות. הגישה המדויקת ביותר לחישוב מצב היסוד לוקחת בחשבון את כל תצורות האלקטרונים האפשריות, אך זה הופך לבלתי ישים עבור מערכות גדולות יותר מכיוון שמספר התצורות גדל אקספוננציאלית עם גודל המערכת.
ה-Handover Iterative Variational Quantum Eigensolver (HI-VQE) הוא שיטה היברידית קוונטית-קלאסית חדשנית לאמידה מדויקת של מצב היסוד של מערכות מולקולריות. הוא משלב חומרה קוונטית עם מחשוב קלאסי, ומשתמש במעבדים קוונטיים כדי לחקור ביעילות תצורות אלקטרוניות מועמדות ומחשב את פונקציית הגל המתקבלת על מחשבים קלאסיים. על ידי יצירת פונקציות גל קומפקטיות אך מדויקות כימית, HI-VQE משפר את המחקר והגילוי בכימיה קוונטית ומדע החומרים.

HI-VQE מפחית את המורכבות החישובית של בעיית המבנה האלקטרוני על ידי אמידה יעילה של מצב היסוד בדיוק גבוה. הוא מתמקד בתת-קבוצה שנבחרה בקפידה של תצורות האלקטרונים הרלוונטיות ביותר, ומייעל גם את הדיוק וגם את היעילות.
בשילוב החוזקות של מחשבים קלאסיים וקוונטיים כאחד, HI-VQE משפר ומדייק באופן איטרטיבי את פונקציית הגל המשוערת הנוכחית. טכניקות בנייה ייחודיות של תת-מרחב מסייעות להפוך את בחירת התצורות ליעילה יותר, כך שלמשתמשים יש שליטה חישובית רבה יותר ודיוק משופר בסימולציות כימיה קוונטית.
אם ברצונך ללמוד על האלגוריתם בעומק רב יותר, תוכל לקרוא את מאמר המחקר הקשור.
תיאור
מספר תצורות האלקטרונים עבור מערכת מולקולרית גדל אקספוננציאלית עם גודל המערכת. עם זאת, עבור מצבים אלקטרוניים מסוימים, כגון מצב היסוד, נפוץ שרק חלק קטן מהתצורות תורמות באופן משמעותי לאנרגיית המצב. שיטות Selected Configuration Interaction (SCI) מנצלות פיזור זה כדי להפחית עלויות חישוביות על ידי זיהוי והתמקדות בתצורות הרלוונטיות ביותר. תת-קבוצה זו של תצורות מכונה תת-מרחב.
HI-VQE ממנף את היעילות הטבועה של מחשבים קוונטיים לייצוג מערכות מולקולריות כדי לסייע בחיפוש תת-המרחב. הוא משלב תת-שגרות קלאסיות וקוונטיות כדי לפתור את בעיית המבנה האלקטרוני בדיוק גבוה. בניגוד לשיטות SCI קוונטיות קיימות, HI-VQE משלב אימון וריאציוני, בנייה איטרטיבית של תת-מרחב, וסינון תצורות מקדים-אלכסוני כדי לשפר את היעילות על ידי הפחתת מדידות קוונטיות, איטרציות ועלויות אלכסון קלאסי. ניתן אפוא להחיל את HI-VQE על מערכות מולקולריות גדולות יותר הדורשות יותר qubit, ומפחית את העלות לפתרון בעיה בגודל נתון לאותה דרגת דיוק.
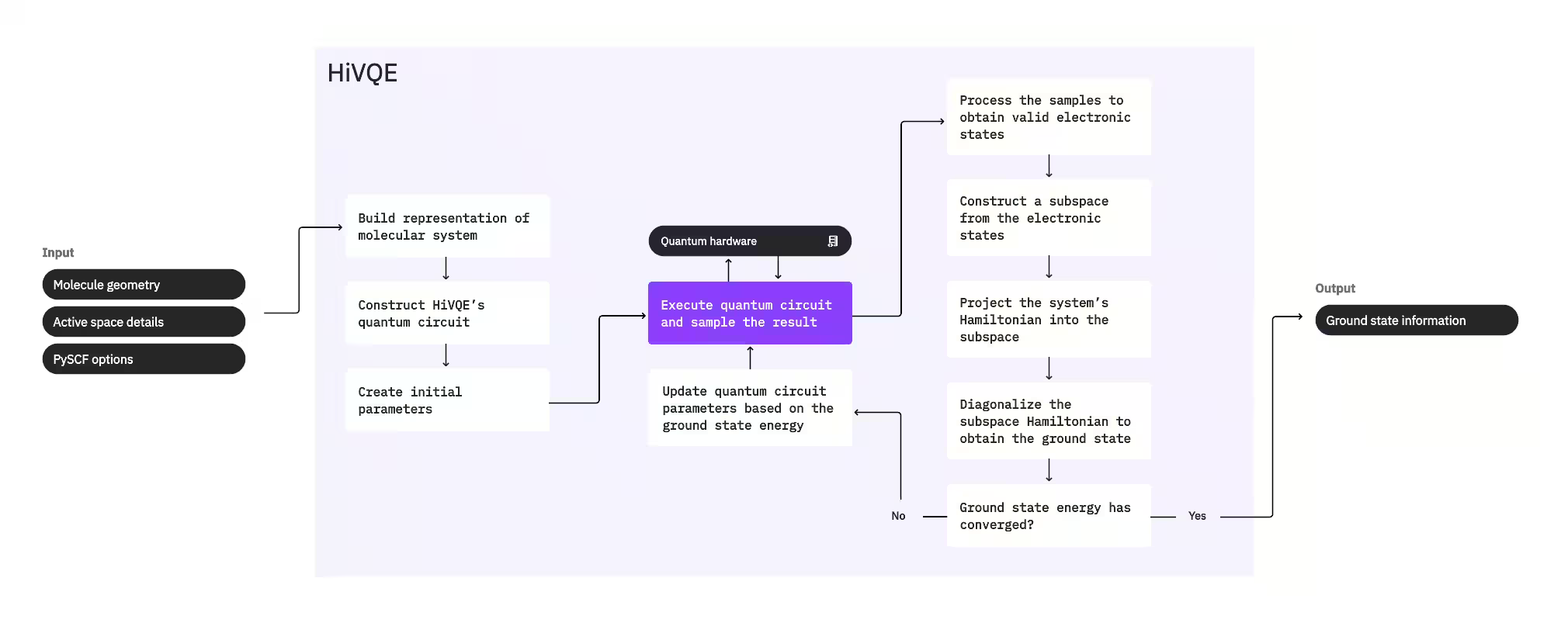
כדי לחשב את מצב היסוד של מערכת, HI-VQE משתמש תחילה בחבילת הכימיה הקלאסית PySCF ליצירת ייצוג מולקולרי מקלטי משתמש, כגון גיאומטריה מולקולרית ומידע מולקולרי אחר. לאחר מכן הוא נכנס ללולאת אופטימיזציה היברידית קוונטית-קלאסית, ומשפר באופן איטרטיבי תת-מרחב כדי לייצג באופן אופטימלי את מצב היסוד תוך מזעור מספר התצורות הכלולות. הלולאה נמשכת עד שמתקיימים קריטריוני התכנסות, כגון גודל תת-מרחב או יציבות אנרגיה, ולאחר מכן פונקציית הגל של מצב היסוד המחושב ואנרגיתו מופקות. ניתן להשתמש בתוצאות אלו לבנייה של עקומות אנרגיה פוטנציאלית מדויקות ולביצוע ניתוח נוסף של המערכת.
לולאת האופטימיזציה מתמקדת בהתאמת פרמטרי Circuit קוונטי ליצירת תת-מרחב איכותי. HI-VQE מציע שלוש אפשרויות Circuit קוונטי: excitation_preserving, efficient_su2, ו-LUCJ. האופטימיזציה מאותחלת קרוב למצב ייחוס Hartree-Fock בשל התאמתו הכללית. ה-Circuit מבוצע לאחר מכן על מכשיר קוונטי ותצורות מדוגמות מהמצב הקוונטי המתקבל לפני שהוחזרו כמחרוזות בינאריות. בשל רעשי מכשיר קוונטי, תצורות מדוגמות מסוימות עשויות להיות פיזיקלית לא חוקיות, ולא לשמר את מספר האלקטרונים או הספין. HI-VQE מטפל בכך באמצעות תהליך שחזור התצורות מחבילת qiskit-addon-sqd, כך שמשתמשים יכולים לתקן תצורות לא חוקיות או להשליך אותן.
לאחר מכן התצורות החוקיות עוברות שלב סינון אופציונלי להסרת אלה שחזויות לתרום באופן מינימלי. זה מפחית את הממד של תת-המרחב, ומוריד בכך את עלות שלב האלכסון. אם הסינון מופעל, נבנה Hamiltonian תת-מרחב ראשוני מהתצורות החוקיות ומבוצע אלכסון עם קריטריוני סיום רופפים מאוד. למרות שדיוק האמפליטודות המתקבלות לכל תצורה נמוך, הוא יעיל לחיזוי אילו תצורות להשאיר מחוץ לתת-המרחב באיטרציה זו, וניתן לחישוב מהיר.
התצורות שנבחרו מתווספות לתת-המרחב, ו-Hamiltonian של המערכת מוקרן לתוך תת-מרחב זה. תת-המרחב מתעדכן באופן איטרטיבי, תוך שמירה על התצורות הרלוונטיות ביותר לאורך איטרציות. גישה זו מנוגדת לשיטות חלופיות מכיוון ש-Circuit הקוונטי אינו צריך לקרב את מצב היסוד המלא בכל שלב.
לאחר מכן, ה-Hamiltonian של תת-המרחב מאולכסן קלאסית לקבלת ערך העצמי הנמוך ביותר ווקטור העצמי המתאים לו, המייצג קירוב של מצב היסוד ואנרגיתו. ככל שאיכות תת-המרחב משתפרת דרך איטרציות, מצב היסוד המחושב מקרב טוב יותר את מצב היסוד האמיתי. ניתן לבצע שלב סינון נוסף בשלב זה להסרת תצורות כלשהן מתת-המרחב שאין להן תרומה משמעותית למצב היסוד המחושב. שלב זה מבטיח שתת-המרחב המועבר לאיטרציה הבאה יהיה קומפקטי ככל האפשר. הדבר מוערך בהתבסס על האמפליטודות שמוחזרות על ידי האלכסון, מכיוון שאלה מייצגות את תרומת החשיבות של כל תצורה למצב היסוד המחושב.
לאחר מכן בדיקת התכנסות קובעת אם אימון נוסף ישפר את התוצאות. אם כן, שלב התרחבות קלאסי אופציונלי מבוצע, פרמטרי Circuit הקוונטי מתעדכנים כדי למזער עוד את האנרגיה המחושבת, והתהליך חוזר. שלב ההתרחבות הקלאסי מייצר תצורות נוספות עבור תת-המרחב, המשלימות את התצורות שנדגמו מהמכשיר הקוונטי. הוא מזהה תחילה את התצורה עם האמפליטודה הגדולה ביותר בתוצאות האלכסון, לפני שמייצר תצורות חדשות עם עירורים בודדים וכפולים מהתצורה שזוהתה. לאחר מכן מספר התצורות הרצוי מאלה מתווסף לתת-המרחב.
לאחר שנקבע שהאיטרציות התכנסו, HI-VQE מחזיר את מצב היסוד המחושב (בצורת המצבים בתת-המרחב ואמפליטודותיהם בפונקציית הגל של מצב היסוד), אנרגיתו, ומדד שונות אנרגיה המעיד אם המצב המחושב מהווה ערך עצמי של ה-Hamiltonian של המערכת.
משתמשים יכולים להחליט על ה-Circuit הקוונטי בשימוש ועל מספר ה-shots שיילקחו לכל Circuit קוונטי, וכן לשלוט בגודל תת-המרחב או לאפשר יצירה קלאסית של תצורות נוספות לסיוע בתצורות שנוצרו קוונטית. בדרך זו משתמשים יכולים להתאים את התנהגות HI-VQE לאפליקציות הרצויות להם.
רישוי
שים לב כי השימוש בפונקציית Qiskit זו מוגבל לבעיות הדורשות לכל היותר 20 qubit, אלא אם כן מתקבל רישיון המעניק מגבלה גבוהה יותר.
שלח אימייל ל-qiskit.support@qunovacomputing.com אם ברצונך לברר אודות קבלת רישיון.
התחלה
ראשית, בקש גישה לפונקציה. לאחר מכן, אמת את זהותך באמצעות מפתח ה-API של IBM Quantum® ובהנחה שכבר שמרת את חשבונך לסביבה המקומית שלך, בחר את פונקציית Qiskit כך:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
דוגמה
הדוגמה הראשונה מראה כיצד לחשב את אנרגיית מצב היסוד עבור מולקולת NH3 באמצעות אלגוריתם HI-VQE.
הגדר את הגיאומטריה המולקולרית והאפשרויות
הגיאומטריה המולקולרית של NH3 מסופקת עם קואורדינטות קרטזיות מופרדות ב-";" לכל אטום.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
ניתן להגדיר ולספק אפשרויות נוספות עבור המערכת המולקולרית בפורמט מילון הבא.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
הרץ את הפונקציה עם קלטי הגיאומטריה והאפשרויות.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
כדאי להדפיס את מזהה עבודת הפונקציה כדי שיהיה ניתן לספקו בבקשות תמיכה אם משהו משתבש.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
דוגמה זו משתמשת אז ב-16 qubit עם 8 אורביטלים של בסיס sto3g עבור מולקולת NH3. בדוק את הסטטוס של עומס עבודה Qiskit Function שלך או קבל תוצאות כך:
print(job.status())
QUEUED
לאחר השלמת העבודה, ניתן לקבל את התוצאות עם מופע result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
כדי לגשת לאנרגיית מצב היסוד, השתמש במפתח "energy". המפתח "eigenvector" מספק את מקדמי ה-CI עם סימון מחרוזת סיביות מתאים של תצורת אלקטרונים המאוחסן עם "states" של התוצאות.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
ביצועים
חלק זה מציג חישובי benchmark מוכחים של HI-VQE עבור מקרה של 24 qubit עבור Li2S, מקרה של 40 qubit עבור מולקולת N2, ומקרה של 44 qubit עבור מערכת FeP-NO.
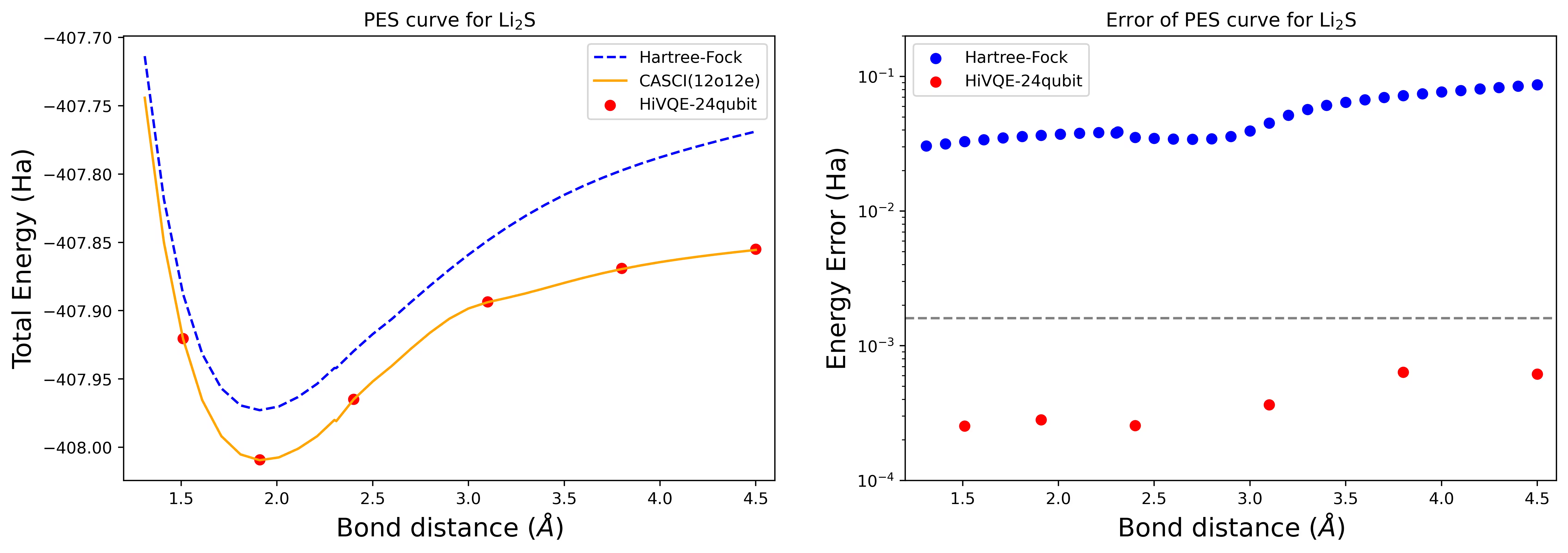
עקומת מישטח אנרגיה פוטנציאלי (PES) לניתוק מולקולת Li2S עם 24 qubit
עקומת PES מוצגת עם ייחוס FCI וניחוש ראשוני מ-RHF, יחד עם שגיאת האנרגיה מייחוס FCI.
 .
.
החישובים בוצעו עם הגיאומטריות והאפשרויות הבאות.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
הנקודות האדומות מייצגות את תוצאות חישוב HI-VQE עבור שש גיאומטריות שונות, ושלוש גיאומטריות המתאימות ל-1.51, 2.40, ו-3.80 אנגסטרום מסופקות כקלט בתא לעיל.
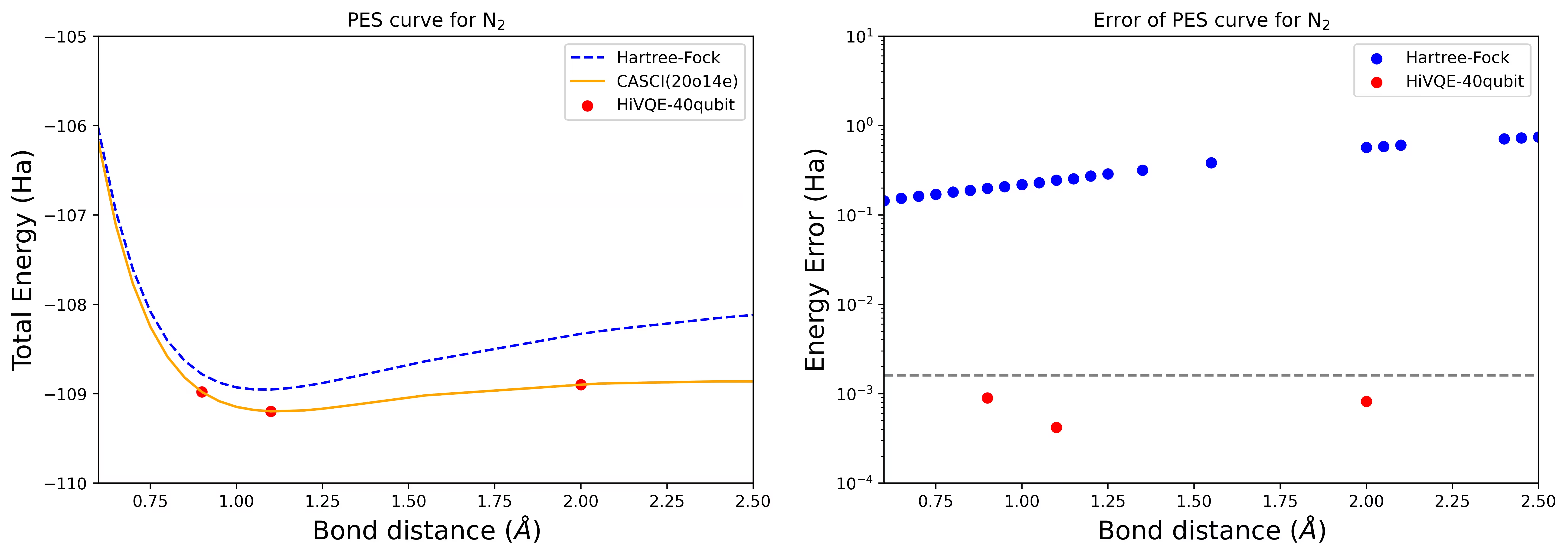
עקומת PES לניתוק עבור מולקולת N2 עם 40 qubit
מולקולת החנקן זוהתה כמערכת multireference עם תרומות אנרגיה קורלציה גדולות מעבר למצב Hartree-Fock. ביצענו חישוב benchmark עבור מולקולת N2 עם בסיס cc-pvdz, (20o,14e) באמצעות בחירת אורביטל פעיל homo-lumo. מספר המרחב הפעיל המלא (CAS) לייצוג בעיה זו הוא 6,009,350,400. אין אפשרות לקבל את פתרון בעיית הערכים העצמיים (עבור אנרגיה ומבנה אלקטרוני) עם מספר מצבים זה באמצעות מחשב שולחני רב-עוצמה (16cpu/64GB). עם HI-VQE, משתמשים יכולים לחפש ביעילות את תת-המרחב של מצבי CAS למציאת תוצאות מדויקות כימית תוך חיסכון משמעותי במשאבי חישוב. הגרפים הבאים מראים את עקומת PES של חישוב HI-VQE בן 40 qubit של ניתוק מולקולת N2.
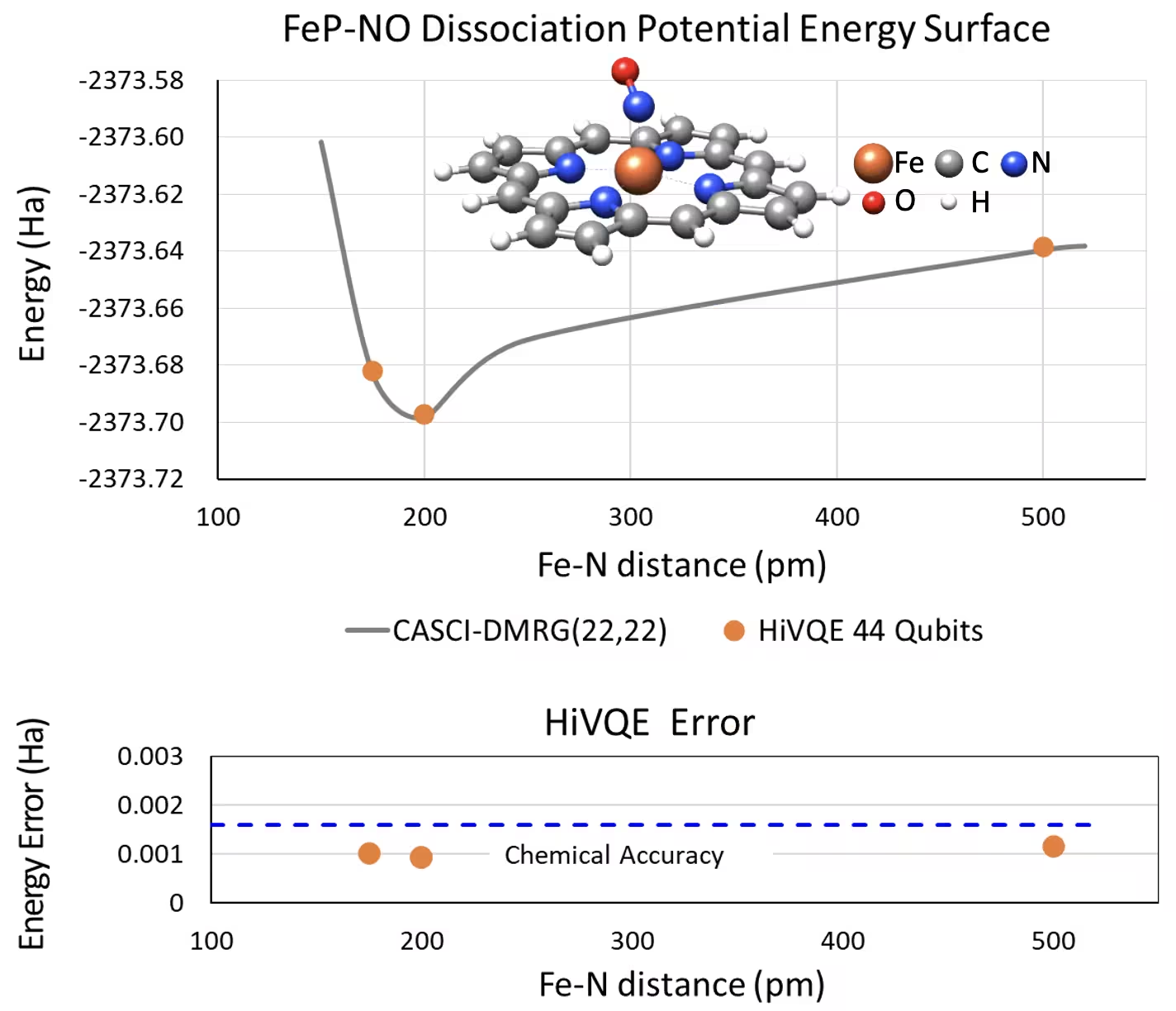
עקומת PES לניתוק עבור ברזל(II)-פורפירין חמישה-מתואם עם מערכת NO עם 44 qubit
מערכת כימית מעניינת נוספת היא קומפלקס ברזל(II)-פורפירין (FeP) עם ליגנד תחמוצת חנקן (NO) מתואם, המייצג מערכת מטלופורפירין רלוונטית ביולוגית הממלאת תפקידים מכריעים בתהליכים פיזיולוגיים שונים. בדוגמה זו, נעשה שימוש ב-HI-VQE להערכת עקומת מישטח אנרגיה פוטנציאלי מדויקת של האינטראקציה הבין-מולקולרית בין FeP ל-NO (אנרגיית מצב היסוד עבור גיאומטריות מופרדות באופן שונה). למערכת המשולבת יש 450 אורביטלים ו-202 אלקטרונים (450o,202e) עם בסיס 6-31g(d) בסך הכל. בחירת אורביטל פעיל homo-lumo שימשה לחישוב המקרה הקטן יותר מהמקרה האמיתי עם (22o,22e). מתוצאות ה-benchmark הבאות, הצלחנו להשיג דיוק כימי (> 1.6 mHa) עם חישוב כימיה קלאסי מתקדם של ייחוס CASCI(DMRG) (22o,22e).
מדדי ביצועים
- גודל המטריצה המדויק הוא מספר הדטרמיננטים לפתרון מדויק, כגון FCI ו-CASCI.
- HI-VQE מדגם ומחשב את תת-המרחב שלו (כלומר, גודל מטריצת HI-VQE).
- הזמן הכולל כולל זמן ריצת QPU ומופעי Qiskit Function עם CPU.
- הדיוק מוערך מהפרש האנרגיה מהפתרון המדויק.
| Chemical system | Number of qubits | Exact matrix size | HI-VQE matrix size | E(diff) from exact (mHa) | Number of iteration | Total time | QPU runtime usage |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
אחזור הודעות שגיאה
אם עומס העבודה שלך נכשל, הסטטוס יהיה ERROR וקריאה ל-job.result() תעלה חריגה:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
קבלת תמיכה
תוכל לשלוח אימייל ל-qiskit.support@qunovacomputing.com לקבלת עזרה בפונקציה זו.
אם אתה זקוק לעזרה בפתרון שגיאה ספציפית, ספק את מזהה עבודת הפונקציה של העבודה שנתקלה בשגיאה.
הצעדים הבאים
- בקש גישה לפונקציה על ידי מילוי טופס זה.
- בקר ב-API reference של פונקציית Qiskit זו.
- נסה את המדריך חישוב עקומת PES לניתוק עבור FeP-NO עם HI-VQE.
- ראה ב-Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- נסה את המדריך עקומות PES לניתוק עם Qunova HiVQE.