דיאגונליזציה קוונטית מבוססת דגימות של המילטוניאן כימי
אומדן זמן שימוש: פחות מדקה על מעבד Heron r2 (הערה: זהו אומדן בלבד. זמן הריצה שלך עשוי להשתנות.)
תוצאות למידה
לאחר מעבר על מדריך זה, משתמשים אמורים להבין:
- כיצד להשתמש בתוסף SQD ל-Qiskit כדי לקרב את אנרגיית מצב היסוד של מערכת מולקולרית באמצעות מחרוזות ביטים שנדגמו מיחידת עיבוד קוונטי (QPU).
- כיצד להשתמש ב-ffsim כדי לבנות מעגל local unitary cluster Jastrow (LUCJ) לסימולציית כימיה קוונטית.
דרישות מוקדמות
אנחנו ממליצים למשתמשים להכיר את הנושאים הבאים לפני מעבר על מדריך זה:
- כימיה קוונטית וקוונטיזציה שניה
- שימוש ב-Sampler primitive לדגימה ממעגלים קוונטיים
רקע
במדריך זה, נראה כיצד לבצע עיבוד-לאחר על דגימות קוונטיות רועשות כדי לקרב את מצב היסוד של מולקולת החנקן באורך קשר שיווי משקל, על ידי שימוש בתוסף SQD ל-Qiskit ליישום אלגוריתם דיאגונליזציה קוונטית מבוססת דגימות (SQD). פרטים נוספים על התוכנה ניתן למצוא בתיעוד המתאים, כולל דוגמה פשוטה כדי להתחיל.
מדריך זה מומלץ למשתמשים המכירים כימיה קוונטית: ספציפית, היכרות עם מציאת אנרגיות מצב היסוד של מולקולה. לסקירה מפורטת של זרימת העבודה, עיין בקורס אלגוריתם הדיאגונליזציה הקוונטית.
SQD היא טכניקה למציאת ערכים עצמיים וווקטורים עצמיים של אופרטורים קוונטיים, כמו המילטוניאן של מערכת קוונטית, על ידי שימוש במחשוב קוונטי וקלאסי מבוזר ביחד. מחשוב קלאסי מבוזר משמש לעיבוד דגימות שהתקבלו ממעבד קוונטי, ולהקרנה ודיאגונליזציה של המילטוניאן יעד בתת-מרחב שהן פורסות. לזרימת עבודה מבוססת SQD יש את השלבים הבאים:
- בחר ansatz מעגלי והחל אותו על מחשב קוונטי על מצב התייחסות (במקרה זה, מצב Hartree-Fock).
- דגום מחרוזות ביטים ממצב קוונטי זה.
- הרץ את תהליך שחזור התצורה העצמי-עקבי על מחרוזות הביטים כדי לקבל את קירוב מצב היסוד.
ידוע ש-SQD עובד היטב כאשר מצב העצמי היעד הוא דליל: פונקציית הגל נתמכת בקבוצה של מצבי בסיס שגודלה אינו גדל באופן אקספוננציאלי עם גודל הבעיה.
כימיה קוונטית
ההמילטוניאן של מערכת מולקולרית ניתן לכתוב כ
כאשר ה- וה- הם מספרים מרוכבים הנקראים אינטגרלים מולקולריים שניתן לחשב מהמפרט של המולקולה באמצעות תוכנית מחשב. במדריך זה, אנחנו מחשבים את האינטגרלים באמצעות חבילת התוכנה PySCF.
לפרטים על האופן שבו ההמילטוניאן המולקולרי נגזר, התייעץ עם ספר לימוד על כימיה קוונטית (לדוגמה, Modern Quantum Chemistry מאת Szabo ו-Ostlund). להסבר ברמה גבוהה על האופן שבו בעיות כימיה קוונטית ממופות למחשבים קוונטיים, בדוק את ההרצאה Mapping Problems to Qubits מ-Qiskit Global Summer School 2024.
אנסץ local unitary cluster Jastrow (LUCJ)
SQD דורש ansatz מעגל קוונטי כדי לשאוב ממנו דגימות. במדריך זה, נשתמש באנסץ local unitary cluster Jastrow (LUCJ) בשל השילוב שלו של מוטיבציה פיזיקלית וידידותיות לחומרה. נשתמש ב-ffsim כדי לבנות את מעגל האנסץ.
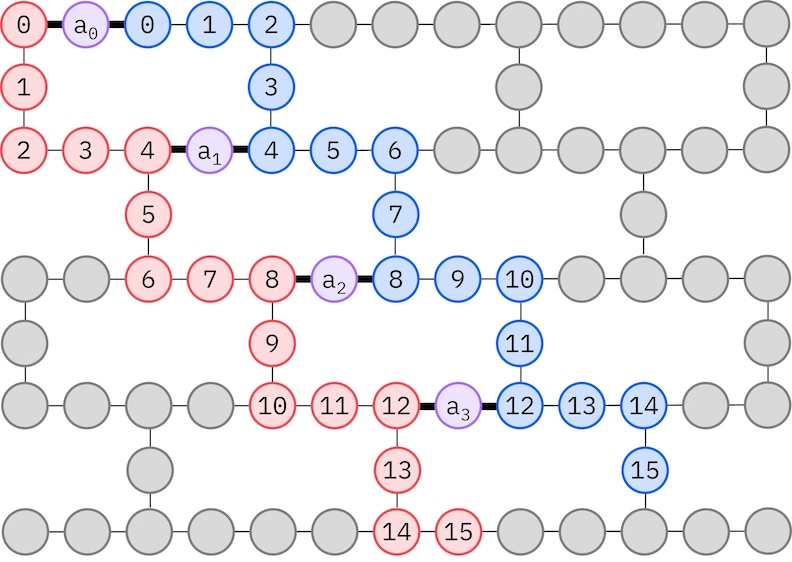
אנסץ ה-LUCJ מתאים את עצמו ל-QPUs עם קישוריות קיוביט מוגבלת. האורביטלים בעלי הספין ממופים לקיוביטים כך שהאנסץ אינו דורש ניתוב עם שערי SWAP. לחומרת IBM® יש טופולוגיית קיוביט של סריג heavy-hex, ובמקרה זה אנחנו יכולים לאמץ תבנית "זיג-זג", המתוארת למטה. בתבנית זו, אורביטלים עם אותו ספין ממופים לקיוביטים עם טופולוגיית קו (עיגולים אדומים וכחולים), וחיבור בין אורביטלים של ספינים שונים קיים בכל אורביטל מרחבי רביעי, כאשר החיבור מתבצע על ידי קיוביט עזר (עיגולים סגולים).
שחזור תצורה עצמי-עקבי
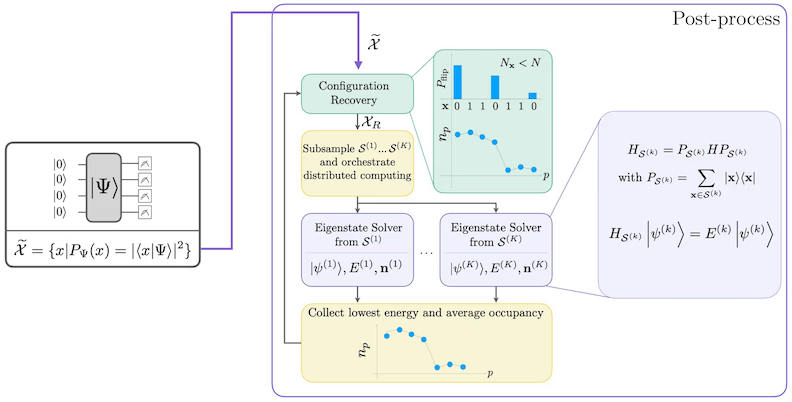
תהליך שחזור התצורה העצמי-עקבי מתוכנן לחלץ כמה שיותר אות מדגימות קוונטיות רועשות. מכיוון שההמילטוניאן המולקולרי שומר על מספר החלקיקים ו-spin Z, הגיוני לבחור ansatz מעגלי ששומר גם על הסימטריות הללו. כאשר מוחלים על מצב Hartree-Fock, המצב המתקבל יש לו מספר חלקיקים קבוע ו-spin Z קבוע בהגדרה נטולת רעש. לכן, חצאי ה-spin- וה-spin- של כל מחרוזת ביטים הנדגמת ממצב זה צריכים להיות בעלי אותו משקל Hamming כמו במצב Hartree-Fock. בשל נוכחות הרעש במעבדים קוונטיים נוכחיים, חלק ממחרוזות הביטים הנמדדות יפרו תכונה זו. צורה פשוטה של בחירה לאחר-מעשה תשליך את מחרוזות הביטים הללו, אבל זה בזבזני מכיוון שמחרוזות ביטים אלה עדיין עשויות להכיל אות כלשהו. תהליך השחזור העצמי-עקבי מנסה לשחזר חלק מהאות הזה בעיבוד-לאחר. התהליך איטרטיבי ודורש כקלט אומדן של התפוסות הממוצעות של כל אורביטל במצב היסוד, שמחושב תחילה מהדגימות הגולמיות. התהליך מורץ בלולאה, וכל איטרציה כוללת את השלבים הבאים:
- עבור כל מחרוזת ביטים המפרה את הסימטריות המוגדרות, הפוך את הביטים שלה עם תהליך הסתברותי המיועד להביא את מחרוזת הביטים קרוב יותר לאומדן הנוכחי של התפוסות האורביטליות הממוצעות, כדי לקבל מחרוזת ביטים חדשה.
- אסוף את כל מחרוזות הביטים הישנות והחדשות שמקיימות את הסימטריות, ודגום תת-קבוצות בגודל קבוע, שנבחר מראש.
- עבור כל תת-קבוצה של מחרוזות ביטים, הקרן את ההמילטוניאן לתוך תת-המרחב שנפרש על ידי וקטורי הבסיס המתאימים (ראה את הסעיף הקודם לתיאור וקטורי הבסיס הללו), וחשב אומדן מצב יסוד של ההמילטוניאן המוקרן על מחשב קלאסי.
- עדכן את האומדן של התפוסות האורביטליות הממוצעות עם אומדן מצב היסוד עם האנרגיה הנמוכה ביותר.
דיאגרמת זרימת עבודה של SQD
זרימת העבודה של SQD מתוארת בדיאגרמה הבאה:
דרישות
לפני תחילת המדריך, ודא שהדברים הבאים מותקנים:
- Qiskit SDK v1.0 ומעלה, עם תמיכה בויזואליזציה
- Qiskit Runtime v0.22 ומעלה (
pip install qiskit-ibm-runtime) - הרחבת Qiskit של SQD v0.11 ומעלה (
pip install qiskit-addon-sqd) - ffsim v0.0.75 ומעלה (
pip install ffsim)
הגדרות
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
דוגמת סימולטור בקנה מידה קטן
במדריך זה, נמצא קירוב למצב היסוד של מולקולת חנקן קרוב למרחק קשר שיווי המשקל שלה. אנחנו משתמשים תחילה בקבוצת בסיס STO-6G קטנה כדי שנוכל לדמות את הניסוי ולוודא שהוא עובד.
שלב 1: מיפוי קלטים קלאסיים לבעיה קוונטית
ראשית, אנחנו מציינים את המולקולה ואת תכונותיה.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
לפני בניית מעגל האנסץ LUCJ, אנחנו מבצעים תחילה חישוב CCSD בתא הקוד הבא. אמפליטודות ו- מחישוב זה ישמשו לאתחול הפרמטרים של האנסץ.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
כעת, אנחנו משתמשים ב-ffsim כדי ליצור את מעגל האנסץ. מכיוון שלמולקולה שלנו יש מצב Hartree-Fock של קליפה סגורה, אנחנו משתמשים בוריאנט מאוזן-ספין של אנסץ ה-UCJ, UCJOpSpinBalanced. אנחנו מגדירים optimize=True בשיטת from_t_amplitudes כדי לאפשר את הפקטוריזציה הכפולה ה"דחוסה" של אמפליטודות ה- (ראה The local unitary cluster Jastrow (LUCJ) ansatz בתיעוד של ffsim לפרטים).
מכיוון שאנסץ ה-LUCJ מתאים את עצמו לקישוריות הזמינה של ה-QPU, עלינו לאתחל את ה-backend של ה-QPU לפני יצירת האנסץ. לעת עתה, ניצור backend גנרי עם מפת הצימוד heavy-hex וסט שערים שאנסץ ה-LUCJ מתפרק אליו באופן טבעי. לאחר מכן, נשתמש ב-ffsim.qiskit.generate_lucj_pass_manager כדי ליצור מנהל מעבר המתמחה בטרנספיילציה של אנסץ ה-LUCJ לה-backend הנתון לפי פריסת ה"זיג-זג" המתוארת בסעיף הרקע על אנסץ LUCJ. פונקציה זו משתמשת בהיוריסטיקת ניקוד כדי למזער את השגיאות הקשורות לפריסה הנבחרת, דבר החשוב אם ה-backend שלך הוא QPU אמיתי, או סימולטור עם מודל רעש. בנוסף להחזרת מנהל המעבר, פונקציה זו מחזירה גם את זוגות הצימוד אלפא-בטא שניתן ליישם על החומרה. אם לא ניתן ליישם את כל הזוגות, היא פולטת אזהרה.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
שלב 2: אופטימיזציה לביצוע חומרה קוונטית
לאחר מכן, אנחנו מבצעים אופטימיזציה של המעגל עבור חומרת יעד. בדרך כלל, שלב זה כולל אתחול ה-backend החומרה ומנהל מעבר עבור ה-backend הזה. אולם, מאחר שאנסץ ה-LUCJ מותאם לקישוריות החומרה, כבר ביצענו פעולות אלה בשלב הקודם. כל שנותר הוא להריץ את מנהל המעבר על המעגל כדי לטרנספייל אותו למעגל ISA שניתן להריץ ישירות על ה-QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
שלב 3: הרצה באמצעות Primitives של Qiskit
לאחר אופטימיזציה של המעגל לביצוע חומרה, אנחנו מוכנים להריץ אותו על חומרת היעד ולאסוף דגימות לאומדן אנרגיית מצב היסוד. מכיוון שיש לנו רק מעגל אחד, נשתמש במצב ביצוע עבודה של Qiskit Runtime ונבצע את המעגל שלנו.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
שלב 4: עיבוד-לאחר והחזרת תוצאה בפורמט קלאסי רצוי
מדד שימושי להערכת איכות הפלט של ה-QPU הוא מספר התצורות התקפות שהוחזרו. תצורה תקפה היא בעלת מספר חלקיקים נכון ו-spin Z נכון, כלומר המחצית הימנית של מחרוזת הביטים בעלת משקל Hamming השווה למספר האלקטרונים בעלי ספין-למעלה, והמחצית השמאלית בעלת משקל Hamming השווה למספר האלקטרונים בעלי ספין-למטה. התא הבא מחשב את שיעור התצורות הדגומות שהן תקפות.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
כל מחרוזות הביטים תקפות מכיוון שאנחנו דוגמים את המעגל על סימולטור ללא רעש. בעת הרצה על QPU רועש, השיעור יהיה קטן מאחד, אך בתקווה יהיה גדול מהשיעור שהיה מצופה אם מחרוזות הביטים היו נדגמות באופן אחיד אקראי, המחושב בתא הבא.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
כעת, אנחנו מעריכים את אנרגיית מצב היסוד של ההמילטוניאן באמצעות הפונקציה diagonalize_fermionic_hamiltonian. פונקציה זו מבצעת את תהליך שחזור התצורה העצמי-עקבי כדי לזקק באופן איטרטיבי את הדגימות הקוונטיות הרועשות כדי לשפר את אומדן האנרגיה. אנחנו מעבירים פונקציית callback כדי שנוכל לשמור את התוצאות הביניים לניתוח מאוחר יותר. ראה את תיעוד ה-API להסברים על הארגומנטים ל-diagonalize_fermionic_hamiltonian.
כאן, אנחנו משתמשים בארגומנט initial_occupancies ל-diagonalize_fermionic_hamiltonian כדי לציין את תצורת Hartree-Fock כניחוש ראשוני לתפוסות האורביטליות במצב היסוד. גישה זו הגיונית עבור מערכות שמצב היסוד שלהן בעל תמיכה משמעותית בתצורת Hartree-Fock, אך היא עשויה שלא להיות מתאימה במצבים אחרים, אם כי שיטות חישוביות מתקדמות יותר עשויות לתת ניחושים ראשוניים טובים יותר במקרים אלה. ציון initial_occupancies מאפשר גם לשחזור התצורה לפעול גם אם לא נדגמו תצורות תקפות, כפי שעשוי לקרות בדגימת מעגל גדול על QPU רועש. ללא ארגומנט זה, שחזור התצורה ייכשל וייזרוק שגיאה אם לא סופקו תצורות תקפות.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
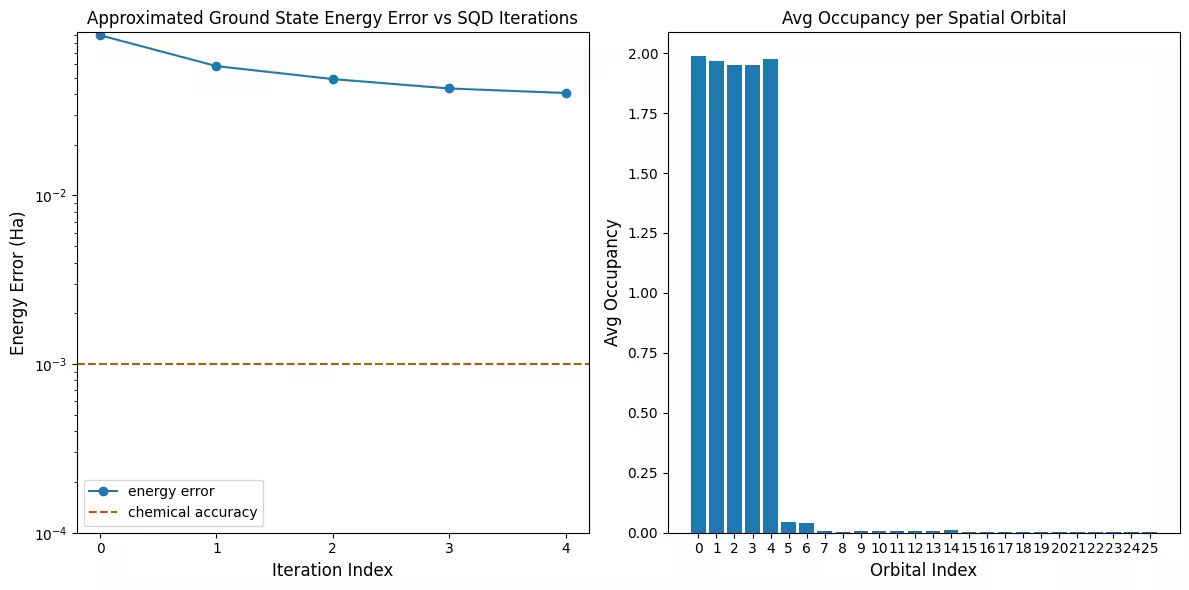
הצגה חזותית של התוצאות
התרשים הראשון מראה שבסימולציה זו אנחנו כבר בטווח של 1 mH מהתשובה המדויקת לאחר האיטרציה הראשונה (דיוק כימי מקובל בדרך כלל להיות 1 kcal/mol 1.6 mH). זוהי מערכת קטנה, אולם, ומכיוון שהדגימות ללא רעש, שחזור התצורה אינו נדרש. על מערכת גדולה יותר המורצת על QPU רועש, ייתכן שיידרשו מספר איטרציות של שחזור תצורה, והדיוק הסופי עשוי להיות גרוע יותר. בדרך כלל, ניתן לשפר את האנרגיה על ידי אפשור יותר איטרציות של שחזור תצורה או הגדלת מספר הדגימות לכל batch.
התרשים השני מראה את התפוסה הממוצעת של כל אורביטל מרחבי לאחר האיטרציה הסופית. אנחנו יכולים לראות שגם האלקטרונים של spin-up וגם של spin-down תופסים את חמשת האורביטלים הראשונים עם הסתברות גבוהה בפתרונות שלנו.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

דוגמת חומרה בקנה מידה גדול
כעת, אנחנו מריצים דוגמה גדולה יותר על חומרה קוונטית אמיתית. כאן, נגזור מרחב פעיל למולקולת החנקן מקבוצת הבסיס cc-pVDZ.
שלבים 1-4
כאן אנחנו מכנסים את כל השלבים יחד לזרימת עבודה אחת בקנה מידה גדול יותר, שמורצת לאחר מכן על חומרה קוונטית אמיתית.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
הצעדים הבאים
אם מצאת עבודה זו מעניינת, ייתכן שתתעניין בחומר הבא:
- דיאגונליזציה קוונטית מבוססת דגימות של Krylov של מודל סריג פרמיוני - מדריך קשור המשתמש במעגלי אבולוציה בזמן במקום ansatz וריאציוני
- שינוי קנה מידה של זרימות עבודה של כימיה SQD עם פותר Dice - דף המראה כיצד להשתמש בתוכנת Dice היעילה יותר לדיאגונליזציה
- תיעוד API של תוסף SQD - עזר לפונקציה
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - המאמר עליו מבוסס מדריך זה